05 July 2022: Articles 
Diagnostic Challenge of Congenital Chloride Diarrhea and Ulcerative Colitis Overlap in an Adult Misdiagnosed with Bartter Syndrome: Case Report and Literature Review
Challenging differential diagnosis, Unusual setting of medical care, Rare disease
Laila Fahad Sadagah123EF*, Ahmad Zaid Makeen12ABEF, Eman Talal Kotbi12EFDOI: 10.12659/AJCR.936715
Am J Case Rep 2022; 23:e936715






Abstract
BACKGROUND: Congenital chloride diarrhea (CCD) is an autosomal recessive disease that is usually diagnosed in early childhood. Mutations in the SLC26A3 gene have been attributed to the primary etiology of disease development. Patients with CCD usually present with electrolyte disturbances, metabolic alkalosis, and chronic diarrhea. Early diagnosis is essential to prevent long-term complications that often require genetic testing. Bartter syndrome is another congenital disorder that has clinical features similar to CCD, which might cause a delay in diagnosis in a few patients.
CASE REPORT: We describe the case of a 28-year-old man who was misdiagnosed as having Bartter syndrome when he was 5 months old based on the clinical features of hypokalemia, metabolic alkalosis, and a family history of Bartter syndrome. He had multiple admissions with diarrhea and was diagnosed with ulcerative colitis. Unfortunately, the course was complicated by renal failure, and the patient underwent a kidney transplant. Persistent metabolic alkalosis with diarrhea after transplantation was unusual in Bartter syndrome. Therefore, his primary diagnosis was challenged and suspicion of CCD was raised, which was confirmed by genetic testing.
CONCLUSIONS: CCD is a rare congenital disorder that requires high clinical suspicion and often a genetic test to confirm diagnosis. Here, we report a patient who was misdiagnosed as having Bartter syndrome until early adulthood owing to several misleading factors. We hope by reporting this case it will raise awareness about CCD in high-prevalence areas and the importance of early diagnosis to prevent serious complications.
Keywords: Bartter Syndrome, SLC26A3 Protein, Human, Kidney Transplantation, Congenital chloride diarrhea, Adult, Alkalosis, Child, Preschool, Colitis, Ulcerative, Diagnostic Errors, diarrhea, Humans, Infant, Male, Metabolism, Inborn Errors
Background
Congenital chloride diarrhea (CCD) is an autosomal recessive disorder caused by a mutation in the
The incidence of CCD appears to have been underestimated. The estimated incidence is approximately 1: 5500 in the Kingdom of Saudi Arabia, mostly because of consanguinity [7]. Finland, Poland, Saudi Arabia, and Kuwait have the highest prevalence rates worldwide [8]. Most patients are diagnosed early, during the first few months of life. Evidence of prenatal polyhydramnios and dilated bowel loops suggests a diagnosis, especially with early onset chronic secretory diarrhea [9]. Delays in the diagnosis ranges from 60 to 217 days, based on the prevalence of the disease between countries [10]; however, few cases are diagnosed even later, during early adolescence or adulthood [7,11,12].
The association between CCD and inflammatory bowel disease is well established; however, the underlying mechanism is not yet completely understood. Downregulation of
Due to phenotypic similarities between CCD and Bartter syndrome, it is challenging to differentiate between them based on clinical features alone, which can result in a delay of diagnosis and appropriate management [15,16]. Genetic testing is often required to confirm the diagnosis of CCD and exclude other conditions.
Herein, we present the case of an adult male with CCD and UC who was misdiagnosed as having Bartter syndrome until early adulthood, which unfortunately complicated his clinical course due to renal failure requiring kidney transplantation.
Case Report
A 28-year-old man was transferred from a pediatric nephrology clinic when he was 14 years old to continue his regular follow-up at the adult nephrology clinic of King Abdulaziz Medical City with a diagnosis of Bartter syndrome. He was diagnosed with Bartter syndrome at the age of 5 months based on the clinical presentation of failure to thrive, volume depletion, and history of antenatal polyhydramnios. These features were accompanied by hypochloremic metabolic alkalosis, hypokalemia, and high urea and creatinine levels. He was started on spironolactone, indomethacin (a nonsteroidal anti-inflammatory drug [NSAID]), and a potassium supplement. His oldest sister was diagnosed as having Bartter syndrome as well, and there was a history of first-degree consanguinity in his parents.
During his childhood, he had multiple admissions to the hospital with electrolyte disturbances, recurrent diarrhea, and abdominal pain. However, he was admitted with a diagnosis of gastroenteritis and was treated symptomatically, requiring electrolyte supplementation at discharge. He was started on growth hormone injections after an endocrinology review for short stature. At 12 years of age, he underwent his first esophagogastroduodenoscopy when he presented with abdominal pain and hematemesis, which revealed a clean-based duodenal ulcer and biopsy-proven
The patient continued to have frequent episodes of watery diarrhea, approximately 8 times per day, with no blood or mucus. Consequently, a colonoscopy was performed for the first time at the age of 13 years. Multiple ulcers were observed from the rectum upward to the cecum with exudation, and the mucosa appeared hyperemic and edematous. Histopathological description indicated active pan-colitis with marked architectural distortion, which confirmed a diagnosis of inflammatory bowel disease-UC (IBD-UC) (Figure 1A, 1B).
Therefore, he was started on pulse steroid induction along with azathioprine as a maintenance therapy. He was followed closely by the gastroenterology team for the diagnosis and subsequent management of IBD, for which he underwent multiple follow-up colonoscopies, and his treatment was escalated to infliximab, followed by vedolizumab over the years. Unfortunately, his diarrhea did not fully resolve with therapy, despite increasing the dose and checking the antibody levels for infliximab.
During early childhood, his clinical course was complicated by renal impairment, which progressively worsened over the years. This was attributed to recurrent episodes of intravascular volume depletion and acute kidney injuries (AKI), secondary to excessive watery diarrhea, and possibly the effect of long-term NSAID use. At the age of 18 years, he was started on intermittent hemodialysis through an arteriovenous fistula for end-stage renal disease. Hemodialysis was continued for 3 years, following which he underwent living non-related kidney transplantation at another institution and was started on immunosuppressive therapy, including prednisolone, mycophenolate mofetil, and tacrolimus (Tables 1 and 2 show the blood, urine, and stool results).
A few years after kidney transplantation, he returned to our institution for regular follow-up with the nephrology and gastroenterology teams. Unfortunately, he had frequent admissions with severe watery diarrhea, causing electrolyte disturbances and AKI, along with metabolic alkalosis, which initially was attributed to severe volume depletion (contraction alkalosis); however, despite the correction of volume depletion, he had persistently elevated bicarbonate levels, with a baseline watery diarrhea 4 to 5 times a day, despite changing mycophenolate mofetil to azathioprine. The remission of UC was also confirmed by several colonoscopies and biopsies, and he was maintained on infliximab to keep his disease inactive.
It was slightly unusual to see features suggestive of Bartter syndrome after renal transplantation, as well as metabolic alkalosis with watery diarrhea. Based on these observations, we challenged the diagnosis of Bartter syndrome and suspected CCD, particularly after reviewing the stool chloride results (Table 3).
To confirm the diagnosis of CCD and rule out Bartter syndrome as an alternative diagnosis, whole exome sequencing was performed using a DNA sample collected from the patient’s peripheral blood, and a homozygous variant c.559G>T P. (Gly187*) in the
Discussion
CCD is a rare autosomal recessive disorder characterized by chronic secretory watery diarrhea with a high fecal chloride content. It can present antenatally or during the early years of life. Similar to other reports and studies, our patient had a history of parental consanguinity, polyhydramnios, failure to thrive, and electrolyte disturbances with metabolic alkalosis [11], which are not the usual findings in diarrhea, as the colonic loss is rich in bicarbonate. However, in CCD, there will be a defect in the Cl–/HCO3– exchanger in the distal ileum and colon, with impaired ability of Cl– absorption and HCO3– secretion, leading to metabolic alkalosis [4], along with intestinal loss of sodium chloride and fluid due to defects in the function of the Na+/H+ exchanger [5], which causes significant volume depletion and AKI.
Diagnosis is usually made in the pediatric age group [1]. Although it can be discovered antenatally, most cases of CCD are diagnosed during infancy [10]. Other studies have reported a median age of diagnosis from late childhood to early puberty. One study reported a diagnosis of CCD in an adult patient, at the age of 22 years, with the presence of a family history of CCD; loss to follow-up contributed to a delayed diagnosis [17]. As in our patient, the diagnosis was confirmed at the age of 28, with multiple possible contributing and confounding factors resulting in this delay (Table 4).
Factors such as similarities in laboratory and clinical findings between CCD and Bartter syndrome, in the form of antenatal findings of polyhydramnios, preterm birth, and failure to thrive during infancy, and hypokalemia and metabolic alkalosis due to hyper-reninemic hyperaldosteronism are found in both disorders. Although CCD presents with watery diarrhea, it is often mistaken for polyuria, which is a common symptom of Bartter syndrome, as the stool is very watery and voluminous, along with daytime/nocturnal enuresis and soiling, which are considered very common findings during early childhood [11]. The fact that our patient’s sister had Bartter syndrome led to further delay in diagnosis, although this was not confirmed by a genetic test since the sister had never been followed up in an adult clinic, did not have diarrhea, and had no complications. The patient’s sister was asymptomatic, except for mild hypokalemia. Moreover, similarities between managing CCD and Bartter syndrome coexist. In CCD, a beneficial effect with improvement of symptoms is seen with the use of prostaglandin synthase inhibitor, which suppresses hyperaldosteronism, and therefore decreases the electrolyte replacement required, which is also compatible with Bartter syndrome. Unfortunately, our patient also had a diagnosis of IBD, for which he was prescribed a proton pump inhibitor as a prophylaxis with steroid induction for IBD-UC. Protein pump inhibitors are known to improve CCD symptoms by decreasing gastric chloride secretion, which further contributed to a delayed diagnosis [18].
The diagnosis of CCD requires careful clinical evaluation, stool chloride analysis, and genetic testing. Furthermore, it is often misdiagnosed as other congenital disorders, such as Bartter syndrome or pseudo-Bartter syndrome, which are known to cause metabolic alkalosis. Although an overlap between CCD and Bartter syndrome can rarely co-occur, as reported in the literature [19], because of clinical similarities, a differential diagnosis of pseudo-Bartter syndrome should be considered and investigated, including diuretic use, laxative abuse, a chronic chloride-deficient diet, cyclical or surreptitious vomiting, excess loss of sodium chloride in sweat due to cystic fibrosis, hypertrophic pyloric stenosis, and CCD [20].
Bartter syndrome is an inherited renal tubular disorder caused by a defective thick ascending limb of the loop of Henle, wherein the patient presents with polyuria, hypokalemia, and metabolic alkalosis [21]. CCD shares clinical and laboratory findings similar to those of Bartter syndrome; hence, patients with CCD might be misdiagnosed as having Bartter syndrome [12,22]. These similarities include the previously mentioned antenatal findings, as well as the laboratory findings, along with frequent episodes of watery diarrhea, which is sometimes misinterpreted by the patient’s caregiver as polyuria. Occasionally, episodes of constipation followed by diarrhea have been reported, mostly because of chronic volume depletion followed by hydration and volume expansion [23].
An association between CCD and IBD has also been reported [24]. A substantial proportion of genetically susceptible patients with CCD, with the
Numerous genetic mutations have been associated with several congenital diarrheal disorders. These might include defects in genes encoding brush border enzymes in cases of lactase and sucrase-isomaltase deficiency, solute carrier and serine protease inhibitor 2 gene mutations in CCD and syndromic congenital sodium diarrhea, respectively, and mutations in the microsomal triglyceride transfer protein gene in cases of familial abetalipoproteinemia [26]. A link between a gain-of-function mutation in the
This mutation has also been noted in cases of classical congenital sodium diarrhea [28].
The development of CCD is associated with more than 100 mutations in
CCD is fatal if not recognized or managed early. Treatment is directed at achieving a positive gastrointestinal balance of the electrolyte by a high intake of chloride, sodium, and potassium. Long-term complications include growth retardation, male sub-/ infertility, and a declining glomerular filtration rate (GFR) [31]. Our patient developed growth retardation and received growth hormone replacement with no significant improvement. Other studies have demonstrated positive growth catch-up after CCD management if patients were diagnosed early and those with a delayed diagnosis did not show normal growth catch-up [7,19].
Declining GFR and developing chronic kidney disease (CKD) are reported in 17.7% [10] of patients with CCD, which is attributable to several factors, including fluid loss and dehydration. Another study reported nephrocalcinosis with hypercalciuria as a possible underlying mechanism [4]. In addition, hypertensive angiopathy has been reported in patients with normotensive CCD, suggesting another mechanism that leads to progressive kidney disease [32]. A progressive decline in the GFR leading to end-stage renal disease requiring renal replacement therapy has been attributed to delayed diagnosis, noncompliance with medication, or inadequate replacement of salts and electrolytes [10]. It is known that chloride and potassium supplementation is beneficial for kidney protection [33]. Although our patient underwent electrolyte replacement, his kidney function continued to deteriorate and progressed to end-stage renal disease, and he underwent kidney transplantation. This finding can be explained by chronic dehydration from longstanding diarrhea caused by CCD, along with episodes of UC flares, leading to further volume depletion and repeated episodes of AKI. A decline in GFR after kidney transplantation in patients with CCD can be explained by the recurrence of nephrocalcinosis in the transplanted kidney and repeated episodes of AKI from volume depletion caused by ongoing severe watery diarrhea. Findings of biochemical features suggestive of Bartter syndrome after kidney transplantation made us challenge the diagnosis, as it is an inherited renal tubular disorder that cannot manifest clinically after renal transplantation; therefore, CCD was suspected, for which genetic testing was performed, confirming the diagnosis.
Conclusions
CCD is a rare disorder that presents both diagnostic and therapeutic challenges. Here, we report a patient with CCD who was misdiagnosed as having Bartter syndrome until adulthood due to voluminous urine, diarrhea, failure to thrive, hypokalemia, and metabolic alkalosis. The diagnosis of UC in our patient complicated the picture and further delayed the diagnosis. The diagnosis of Bartter syndrome in our patient was challenging due to the presence of chronic kidney disease, persistent watery diarrhea, electrolyte disturbances, and metabolic alkalosis after kidney transplantation. In our patient, the diagnosis of Bartter syndrome in the older sister was also unfortunately misleading. We hope that this case report will increase awareness of the diagnosis of CCD among physicians, especially in areas with high disease prevalence.
Tables
Table 1.. Sample of the patient’s blood laboratory results, before and after hydration.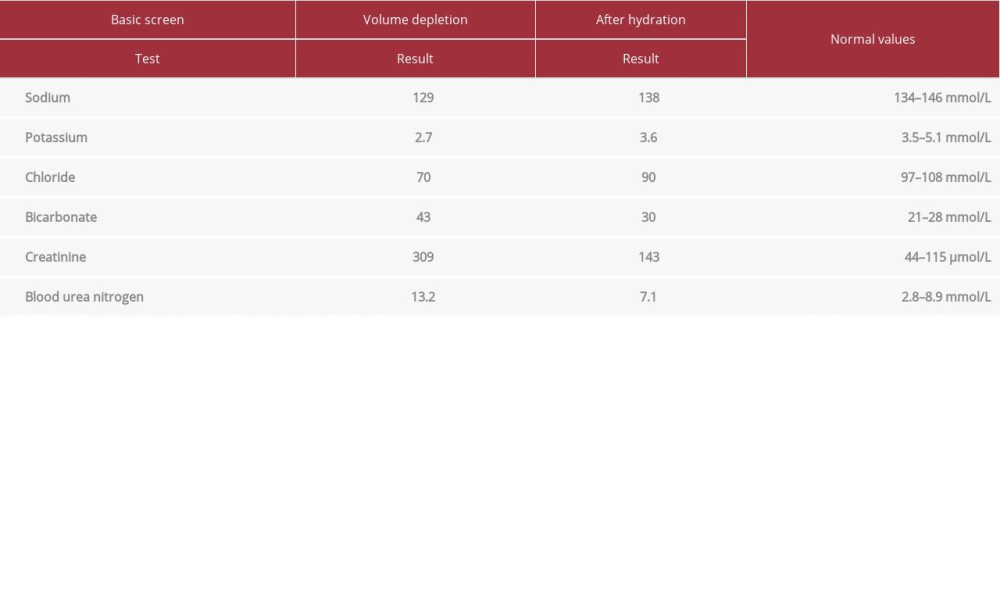 Table 2.. Urine and stool test results.
Table 2.. Urine and stool test results.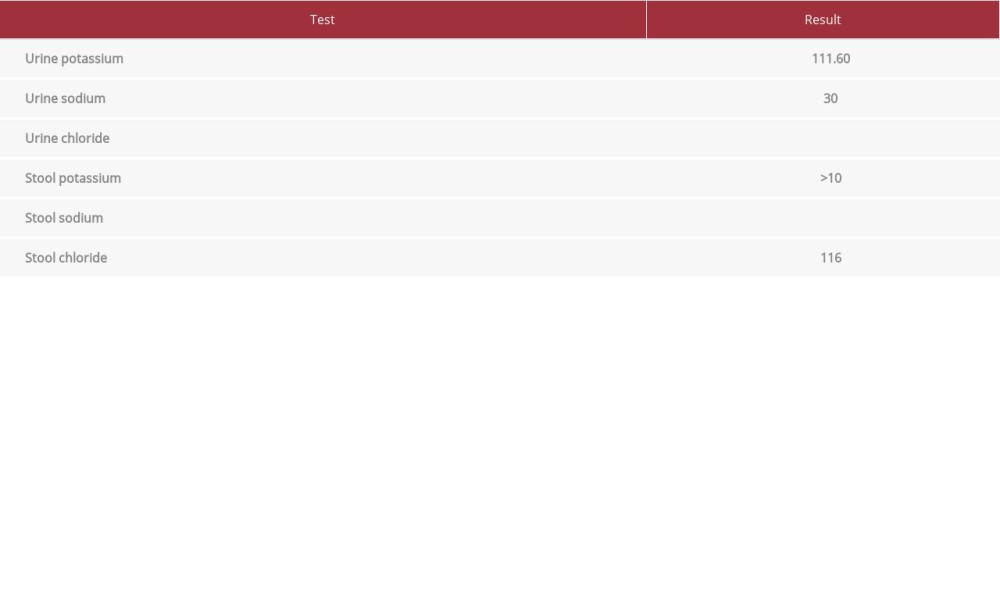 Table 3.. Stool chloride level always exceeded 90 mmol/L, which supported the diagnosis of congenital chloride diarrhea.
Table 3.. Stool chloride level always exceeded 90 mmol/L, which supported the diagnosis of congenital chloride diarrhea.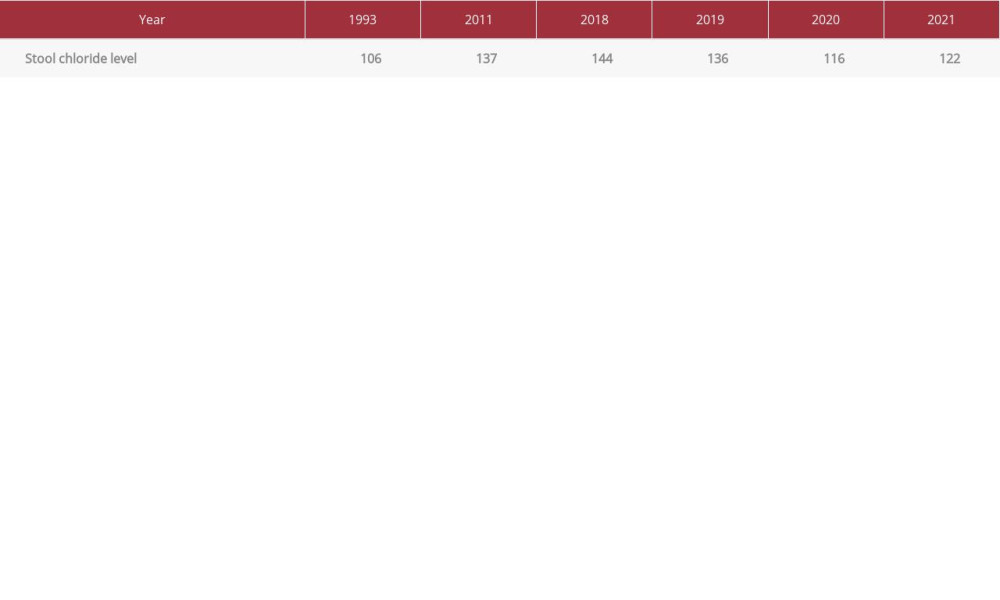 Table 4.. Summary of the literature review findings of similar cases of congenital chloride diarrhea.
Table 4.. Summary of the literature review findings of similar cases of congenital chloride diarrhea.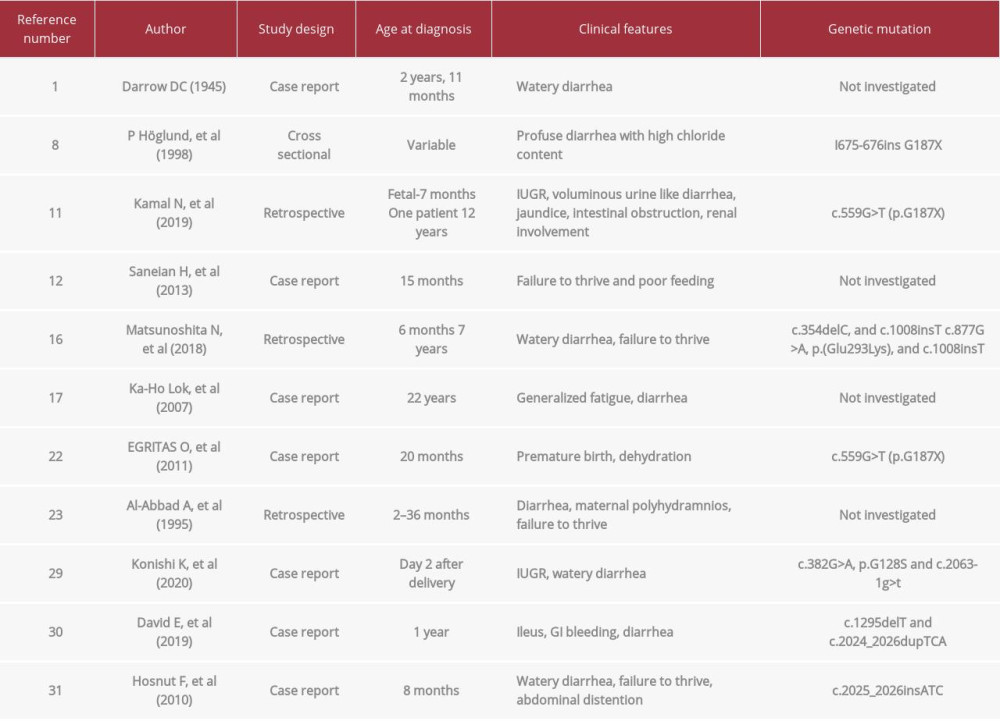
References:
1.. Wedenoja S, Hã-Glund P, Holmberg C, Review article: The clinical management of congenital chloride diarrhoea: Aliment Pharmacol Ther, 2010; 31(4); 477-85
2.. Darrow DC, Congenital alkalosis with diarrhea: J Pediatr, 1945; 26(6); 519-32
3.. Gamble JL, Fahey KR, Appleton J, MacLachlan E, Congenital alkalosis with diarrhea: J Pediatr, 1945; 26(6); 509-18
4.. Wedenoja S, Örmälä T, Berg UB, The impact of sodium chloride and volume depletion in the chronic kidney disease of congenital chloride diarrhea: Kidney Int, 2008; 74(8); 1085-93
5.. Musch MW, Arvans DL, Wu GD, Chang EB, Functional coupling of the downregulated in adenoma Cl–/base exchanger DRA and the apical Na+/ H+ exchangers NHE2 and NHE3: Am J Physiol Gastrointest Liver Physiol, 2009; 296(2); G202-10
6.. Holmberg C, Electrolyte economy and its hormonal regulation in congenital chloride diarrhea: Pediatr Res, 1978; 12(2); 82-86
7.. Shawoosh HA, Badgaish F, Raboie E, Basrawi T, Congenital chloride diarrhea from the west coast of the Kingdom of Saudi Arabia: Saudi Med J, 2000; 21(2); 207-8
8.. Höglund P, Auranen M, Socha J, Genetic background of congenital chloride diarrhea in high-incidence populations: Finland, Poland, and Saudi Arabia and Kuwait: Am J Hum Genet, 1998; 63(3); 760-68
9.. Poggiani C, Pasinetti G, Auriemma A, Menghini P, [A case of congenital chloridorrhea: The diagnostic contribution of pre- and postnatal echography.]: Pediatr Med Chir, 1992; 14(5); 557-58 [in Italian]
10.. di Meglio L, Castaldo G, Mosca C, Congenital chloride diarrhea clinical features and management: A systematic review: Pediatr Res, 2021; 90(1); 23-29
11.. Kamal NM, Khan HY, El-Shabrawi MHF, Sherief LM, Congenital chloride losing diarrhea: Medicine, 2019; 98(22); e15928
12.. Saneian H, Bahraminia E, Congenital chloride diarrhea misdiagnosed as pseudo-Bartter syndrome: J Res Med Sci, 2013; 18(9); 822-24
13.. Ding X, Li D, Li M, Tian D, Tumor necrosis factor-α acts reciprocally with solute carrier family 26, member 3, (downregulated-in-adenoma) and reduces its expression, leading to intestinal inflammation: Int J Mol Med, 2018; 41(3); 1224-32
14.. Asano K, Matsushita T, Umeno J, A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population: Nat Genet, 2009; 41(12); 1325-29
15.. Wedenoja S, Pekansaari E, Höglund P, Update on SLC26A3 mutations in congenital chloride diarrhea: Hum Mutat, 2011; 32(7); 715-22
16.. Matsunoshita N, Nozu K, Yoshikane M, Congenital chloride diarrhea needs to be distinguished from Bartter and Gitelman syndrome: J Hum Genet, 2018; 63(8); 887-92
17.. Lok KH, Hung HG, Li KK, Li KF, Szeto ML, Congenital chloride diarrhea: A missed diagnosis in an adult patient: Am J Gastroenterol, 2007; 102(6); 1329-30
18.. Aichbichler BW, Zerr CH, Santa Ana CA, Proton-pump inhibition of gastric chloride secretion in congenital chloridorrhea: N Engl J Med, 1997; 336(2); 106-9
19.. Sakallı H, Bucak H, Type IV neonatal Bartter syndrome complicated with congenital chloride diarrhea: Am J Case Rep, 2012; 13; 230-33
20.. Matsunoshita N, Nozu K, Shono A, Differential diagnosis of Bartter syndrome, Gitelman syndrome, and pseudo-Bartter/Gitelman syndrome based on clinical characteristics: Genet Med, 2016; 18(2); 180-88
21.. da Silva Cunha T, Heilberg IP, Bartter syndrome: Causes, diagnosis, and treatment: Int J Nephrol Renovasc Dis, 2018; 11; 291-301
22.. Eğrıtaş O, Dalgiç B, Wedenoja S, Congenital chloride diarrhea misdiagnosed as Bartter syndrome: Turk J Gastroenterol, 2011; 22(3); 321-23
23.. Al-Abbad A, Nazer H, Sanjad SA, Al-Sabban E, Congenital chloride diarrhea: A single center experience with ten patients: Ann Saudi Med, 1995; 15(5); 466-69
24.. Norsa L, Berni Canani R, Duclaux-Loras R, Inflammatory bowel disease in patients with congenital chloride diarrhoea: J Crohn Colitis, 2021; 15(10); 1679-85
25.. Kim ES, Song JS, Ki CS, Choe YH, Kang B, Development of Crohn’s disease in a child with SLC26A3-related congenital chloride diarrhea: Report of the first case in East Asia and a novel missense variant: Ann Lab Med, 2021; 41(2); 255-57
26.. Terrin G, Tomaiuolo R, Passariello A, Congenital diarrheal disorders: An updated diagnostic approach: Int J Mol Sci, 2012; 13(4); 4168-85
27.. Fiskerstrand T, Arshad N, Haukanes BI, Familial diarrhea syndrome caused by an activating GUCY2C mutation: N Engl J Med, 2012; 366(17); 1586-95
28.. van Vugt AHM, Bijvelds MJC, de Jonge HR, A potential treatment of congenital sodium diarrhea in patients with activating GUCY2C mutations: Clin Transl Gastroenterol, 2021; 12(11); e00427
29.. Konishi K, Mizuochi T, Honma H, A novel de novo SLC26A3 mutation causing congenital chloride diarrhea in a Japanese neonate: Mol Genet Genomic Med, 2020; 8(11); e1505
30.. Dávid É, Török D, Farkas K, Genetic investigation confirmed the clinical phenotype of congenital chloride diarrhea in a Hungarian patient: A case report: BMC Pediatr, 2019; 19(1); 16
31.. Özbay Hoşnut F, Karadağ Öncel E, Öncel MY, Özcay F, A Turkish case of congenital chloride diarrhea with SLC26A3 gene (c.2025_2026insATC) mutation: Diagnostic pitfalls: Turk J Gastroenterol, 2010; 21(4); 443-47
32.. Pasternack A, Perheentupa J, Hypertensive angiopathy in familial chloride diarrhea: Lancet, 1967; 289(7493); 793
33.. Hihnala S, Höglund P, Lammi L, Long-term clinical outcome in patients with congenital chloride diarrhea: J Pediatr Gastroenterol Nutr, 2006; 42(4); 369-75
Tables
Table 1.. Sample of the patient’s blood laboratory results, before and after hydration.Table 2.. Urine and stool test results.Table 3.. Stool chloride level always exceeded 90 mmol/L, which supported the diagnosis of congenital chloride diarrhea.Table 4.. Summary of the literature review findings of similar cases of congenital chloride diarrhea.Table 1.. Sample of the patient’s blood laboratory results, before and after hydration.Table 2.. Urine and stool test results.Table 3.. Stool chloride level always exceeded 90 mmol/L, which supported the diagnosis of congenital chloride diarrhea.Table 4.. Summary of the literature review findings of similar cases of congenital chloride diarrhea. In Press
06 Mar 2024 : Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.942937
12 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943244
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943275
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943411
Most Viewed Current Articles
07 Mar 2024 : Case report 
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133
10 Jan 2022 : Case report 
DOI :10.12659/AJCR.935263
Am J Case Rep 2022; 23:e935263
19 Jul 2022 : Case report
DOI :10.12659/AJCR.936128
Am J Case Rep 2022; 23:e936128
23 Feb 2022 : Case report
DOI :10.12659/AJCR.935250
Am J Case Rep 2022; 23:e935250