17 November 2023: Articles 
Erdheim-Chester Disease with Renal Mass Presentation: Report of the First Case From Palestine and a Review of the Literature
Rare disease
Hanood Bassam Abu Rass1ABCDEF*, Mohammad Abuawad2DEF, Yazan Abueideh3ABCDF, Ezzideen Luai Malhis4ABCDEFDOI: 10.12659/AJCR.941912
Am J Case Rep 2023; 24:e941912






Abstract
BACKGROUND: Erdheim-Chester disease (ECD), a form of non-Langerhans-cell histiocytosis, is extremely rare. The mean age of individuals with ECD is in their 50s. Histiocytic infiltration of vital organ systems is a potential cause of substantial morbidity, which is associated with the multisystemic form of ECD. This report presents the first case of ECD with renal abnormalities in Palestine.
CASE REPORT: A 54-year-old woman with no medical or surgical history presented with 6 months of bilateral flank pain with no radiation or fever. A physical examination revealed only bilateral flank pain. Urine tests showed microhematuria. Laboratory test results showed increased serum creatinine levels (1.21 mg/dL) and microcytic anemia. A CT scan revealed significant multi-organ abnormalities, including renal abnormalities with a hairy kidney sign, pericardial effusion, and an osteolytic lesion of the spine. The hairy kidney sign is pathognomonic for ECD, so the renal mass was biopsied to confirm the diagnosis. The biopsy showed foamy histiocytes, lymphocytes, and plasma cells. Foamy histiocytes were CD68-positive and negative for S100, CD1a, and HMB45. PAx5 and CD3 immunostaining showed T-predominant B-lymphocyte mixtures.
CONCLUSIONS: In the setting of systemic symptoms and imaging abnormalities such as presence of the hairy kidney sign, pericardial effusion, and osteolytic lesion of the spine, it is necessary to examine the possibility of ECD and proceed with a biopsy for confirmation. This is the first case in Palestine to be reported and the second case worldwide with a renal mass as an atypical presentation.
Keywords: Histiocytosis, Flank Pain, Erdheim-Chester Disease, Female, Humans, Middle Aged, Pericardial Effusion, Biopsy, Tomography, X-Ray Computed
Background
Xanthoma is a lesion that results from the phagocytizing of lipids by macrophages forming foamy cells [1]. In the past, it was known that this lesion could only occur on the skin in patients with hypercholesteremia [2]. However, it was later discovered that xanthomas also appear in other organs due to different inborn errors in the metabolism of lipids inside cells, such as Gaucher and Niemann-Pick disease [3]. In 1930, 2 cases were published of people who died as a consequence of heart failure, in which deposition of foamy histiocytes with lipids in the lungs, heart, liver, and bones was discovered. It was believed to be different from Langerhans-histiocytes, in which the foamy histiocytes do not contain lipids. This disease was called Erdheim-Chester disease (ECD) in honor of the 2 scientists who first described it [4].
The fact that only 1500 cases of the disease have been reported by the year 2020 since its discovery demonstrates its rarity [5]; 70% of the affected patients were males in their 50s [6,7]. Thanks to the development of new diagnostic techniques, BRAFV600E mutation was detected in ECD via pyrosequencing of biopsy samples [8]. The discovery of this mutation in CD34+ hematopoietic cells suggests that the origin of ECD cells is the bone marrow [9]. The deposition of lipohistiocytes in various organs can induce an inflammatory reaction that plays a major role in the progression of fibrotic lesions [10]. In addition, ECD cells express a marker called p16Ink4a, which contributes to the recruitment of immune cells with specific cytokines [11]. Long-bone osteosclerotic lesions [7], cerebral involvement [12], periaortic infiltration [6], perirenal infiltration [7], xanthelasma-like lesions [13], and various endocrine deficiencies are commonly seen in this disease [14]. The distinguishing histopatho-logical features of ECD are foamy histiocytes with surrounding lymphocytes and fibrosis. Immunohistochemistry shows CD68 and CD163 are positive, but S100 is rarely positive [15]. Because the BRAFe600 mutation is present in 50–60% of ECD cases [16], administration of a BRAFs600 inhibitor is recommended [5].
Case Report
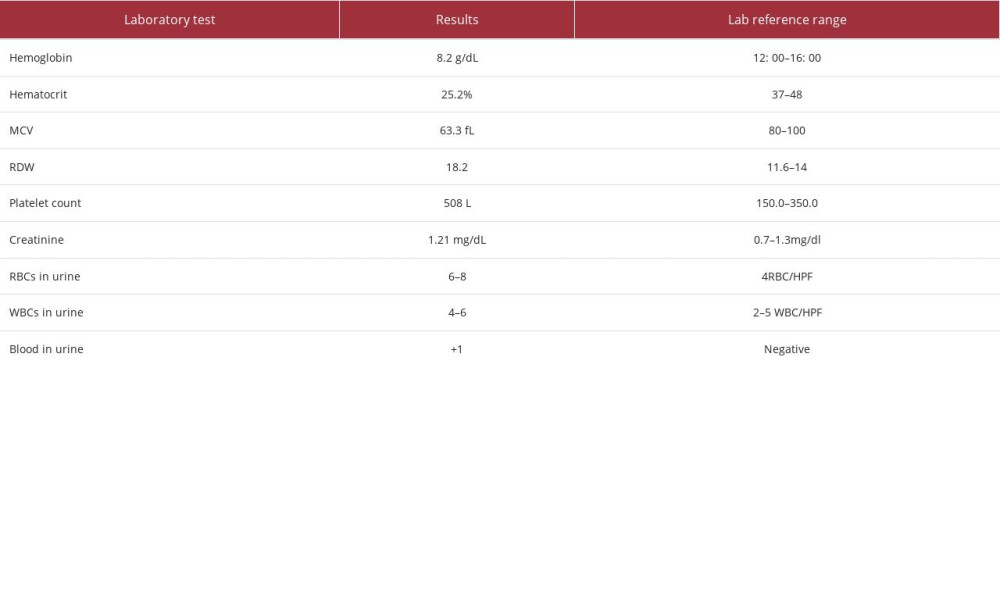
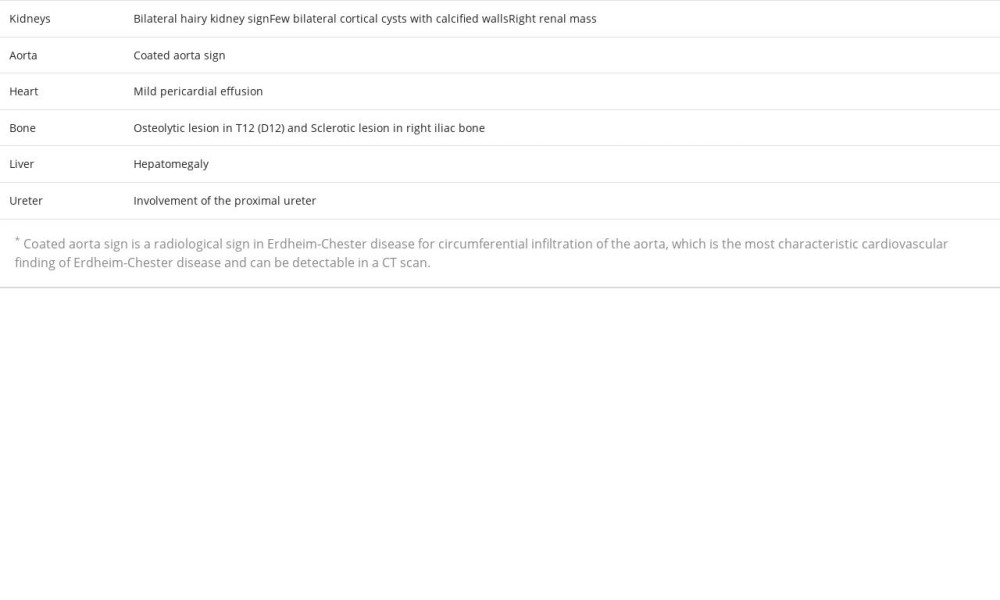
A 54-year-old woman with no medical or surgical history presented to the Emergency Department with 6 months of bilateral flank pain, which was dull, occurred gradually, and fluctuated in intensity, but it did not radiate anywhere and was not associated with a fever. The pain was 5/10 on the visual analog scale. Her history was significant for generalized fatigue, anorexia, and weight loss of more than 10% within the last 6 months, but she had no night sweats, hematuria, dysuria, or dyspnea. This is the first time that the patient had sought medical advice since the beginning of her symptoms. Bilateral flank tenderness was the only significant physical examination finding. For evaluation, the patient was asked to give a urine sample, and blood was drawn. The urine analysis showed microhematuria, and her laboratory test results showed elevated creatinine levels and microcytic anemia (Table 1). Ultrasound imaging showed substantial abnormal findings of both kidneys (Table 2). On the next day, a CT scan of the chest, abdomen, and pelvis was done and exhibited various changes in many organs (Figures 1–3). Table 3 displays the abnormal CT findings. A CT scan revealed a faint hypodense lesion in D12 with a sclerotic rim (Figure 1). A sclerotic lesion on the right iliac bone was also seen, as well as a round non-enhancing lesion in the cortex of the right kidney, which shows a dense (calcified) wall measuring 38 HU in density (Figure 3). The soft tissue covering the kidney extended to also cover the proximal part of the ureters and caused the uro-mechanical obstruction. Ultrasound imaging (not shown)showed that the right kidney appeared heterogeneous and bulky with loss of corticomedullary differentiation, and also revealed a round echo-genic lesion that was 3.2 cm in diameter.
As hairy kidney sign (Figure 3) is pathognomonic for ECD (19), this diagnosis was suspected, and a biopsy from the renal mass was taken for confirmation. It showed an infiltrate of foamy histiocytes admixed with scattered lymphocytes and plasma cells (Figure 4). The foamy histiocytes were positive for CD68 (Figure 5) and negative for S100, CD1a, and HMB45. Mixtures of B and T lymphocytes with a predominance of T lymphocytes were seen and were confirmed by PAx5 and CD3 immunostaining.
Discussion
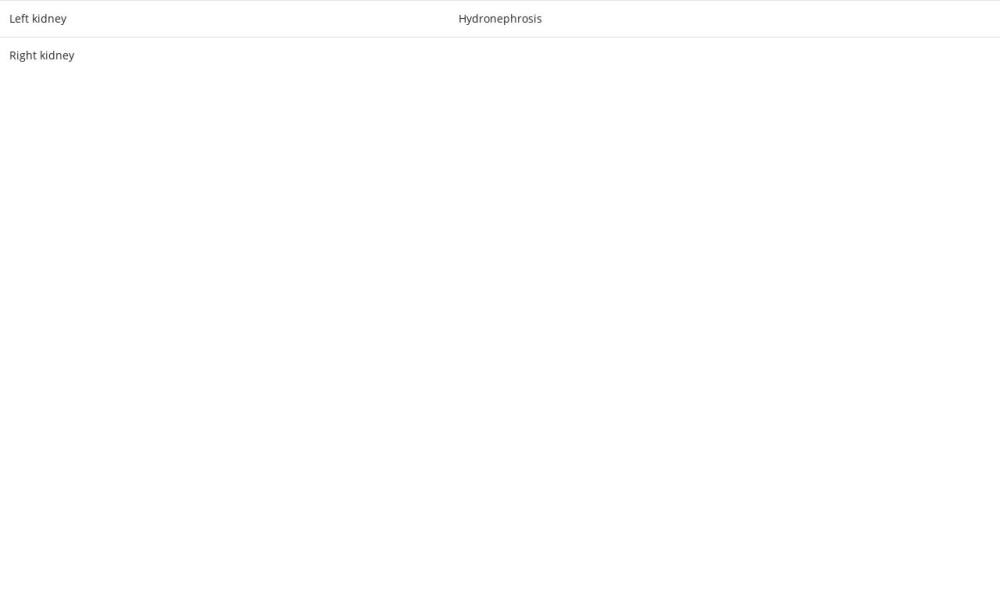
To the best of our knowledge, this is the first case of ECD to be reported in Palestine. The perirenal infiltration and the thickening of the renal septa explain the selection of the term “ hairy” to describe the CT scan finding [15]. Despite the fact that retroperitoneal fibrosis is the most common cause of ureteral obstruction in ECD, our patient had hydronephrosis in the left kidney due to involvement in the proximal ureteral part, without any evidence of fibrosis [17]. A retrospective study of 24 ECD patients with different urological manifestations revealed that the proximal ureter was involved in 14 cases [18]. Moreover, the existence of a solid renal mass resembling renal cell carcinoma was only reported once before [17]. Based on this, we infer that our patient is the second reported case of a renal mass in ECD.
The microcytic anemia can be explained by the fact that ECD induces secretion of systemic inflammatory markers [10], but hematological manifestations should not always be attributed to this cause, as they can designate the presence of a myeloid neoplasm, especially when lymphocytosis or thrombocytosis are present [19]. There are other causes of microcytic anemia, including nutritional deficiency, hereditary conditions, acute or chronic bleeding, and chronic disease. Unfortunately, no blood tests were performed for iron studies for our patient. ECD is considered an inflammatory myeloid neoplasm. Approximately 10% exhibit a correlation with myeloproliferative neoplasms and/or myelodysplastic syndromes. Several notable manifestations of ECD include the high prevalence of long bone involvement (80–95%), the presence of a coated aorta in around 40% of cases, pericardial effusion occurring in 24% of patients, and pseudo-tumoral infiltration of the right atrium observed in 36% of individuals affected by the disease [20]. Our patient’s platelet count was 503 L. Consequently, it was advisable to obtain a bone marrow biopsy.
The deposition of fat around the aorta is one of the distinctive signs that have been seen in numerous patients with ECD. The periaortic infiltration can extend along the aorta and its branches [5]. In our patient, the periaortic infiltration had expanded from the lower part of the thoracic aorta to the abdominal bifurcation. This should not be underestimated, as a case of ECD with ruptured abdominal aortic aneurysm with successive retroperitoneal fibrosis was previously reported; the diagnosis of ECD was made 4 years later after developing periaortic infiltration in the thoracic aorta [21], suggesting that such patients should be warned of the risk of aortic rupture due to the progressive precipitation of fatty tissue around the aorta and the inflammation it induces [22].
Pericardial effusion is one of the represented findings of ECD in the literature [23]. Several cases of ECD with pericardial effusion have been published [24,25]. A complication that must not be misjudged is the rapid accumulation of a large amount of fluid, leading to cardiac tamponade [26]. In addition to this, 40% of ECD patients with cardiac manifestations show a pseudotumor in the right atrium, which is revealed by cardiac MRI [7]. Therefore, a cardiac MRI was mandatory for our patient.
The involvement of the appendicular skeleton with osteosclerotic lesions is more common than involvement of the axial skeleton [7]. Therefore, an osteoblastic lesion in the right iliac bone is considered to be an uncommon ECD finding. Unique findings in our patient include the axial skeleton involvement and the osteolytic lesion seen in the vertebral body of T12. These osteolytic lesions have been described in the literature reviews of a few ECD case reports [27–30]. There have been 2 case reports of axial osteolytic lesions presenting with multiple liver nodules, which is a special characteristic of the disease that was first described in 2003 [27,31].
The biopsy taken from the renal mass showed an infiltrate of foamy histiocytes admixed with scattered lymphocytes and plasma cells, which fits the typical findings for ECD found in the literature [13]. Further testing was done for the clusters of differentiation (CDs) of cell surface markers. The foamy histiocytes were positive for CD68, which helps to categorize cells of macrophage lineages such as histiocytes and multinucleated giant cells [32]. The biopsy was negative for S100 and CD1a surface molecules, which are characteristically positive in Langerhans cell histiocytosis (LCH), which is a rare histiocytic condition that causes clonal development of neoplastic Langerhans cells, but negative in ECD [33]. The HMB45 surface molecule was negative. This immunohistochemistry test should be done for any renal mass with the above-mentioned morphological characteristics to rule out xanthogranulomatous pyelonephritis and perivascular epithelioid cell tumors (PECOMAs). Xanthogranulomatous pyelonephritis is an extremely rare condition brought on by a chronically obstructive pathological calculus or infection. The renal parenchyma is destroyed by a protracted but incomplete immune response and lipid-laden macrophages, which were not present in our patient. Loss of renal outline and paradoxical constriction of the renal pelvis are the most prevalent radiographic findings. The enlarged calyces appear multiloculated, like a bear’s paw print [34]. In our case, radiological findings revealed bilateral enlargement of both kidneys, without stones. PECOMAs are a family of related mesenchymal neoplasms that include lymph-angiomyomatosis tumors, angiomyolipoma, clear cell lung tumor, and several other tumors that occur in the visceral and soft tissues. Pathologically, they form nests of usually epithelioid and sometimes spindle cells with clear to granular eosinophilic cytoplasm and a focal association with the blood vessel wall [35]. Mixtures of B and T lymphocytes with predominance of T lymphocytes were seen, and further clusters of differentiation testing were positive for PAx5 and CD3 immunostaining. The last finding is consistent with the fact that an inflammatory reaction could be initiated in organs with lipohistiocyte deposition, which then causes progression of fibrosis [10].
Conclusions
A 54-year-old woman came to the ER with bilateral flank pain with microcytic anemia and an increased serum creatinine level. A CT scan showed hairy kidney sign, which is pathognomonic for ECD, in addition to pericardial effusion, and an osteolytic lesion of the spine. The biopsy confirmed the diagnosis. The presentation of the disease was very indolent and insidious. When these symptoms and imaging abnormalities are present, physicians should obtain a biopsy to rule out ECD. To the best of our knowledge, this is the first case of ECD in Palestine to be reported and the second reported case of a renal mass worldwide.
Figures
References:
1.. Zak A, Zeman M, Slaby A, Vecka M, Xanthomas: Clinical and pathophysio-logical relations: Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub, 2014; 158(2); 181-88
2.. Arning E, Lippmann A, [Essential cholesterolemia with xanthoma formation.]: Z klin Med, 1920; 89; 107-19 [in German]
3.. Bloom W, Splenomegaly (Type Gaucher) and lipoid-histiocytosis (Type Niemann): Am J Pathol, 1925; 1(6); 595-626.9
4.. Chester W, [About lipoid granulomatosis.]: Virchows Arch path Anat, 1930; 279(2); 561-602 [in German]
5.. Papo M, Emile J-F, Maciel TT, Erdheim-Chester disease: A concise review: Curr Rheumatol Rep, 2019; 21(12); 66
6.. Cohen-Aubart F, Emile J-F, Carrat F, Phenotypes and survival in Erdheim-Chester disease: Results from a 165-patient cohort: Am J Hematol, 2018; 93(5); E114-17
7.. Estrada-Veras JI, O’Brien KJ, Boyd LC, The clinical spectrum of Erdheim-Chester disease: An observational cohort study: Blood Adv, 2017; 1(6); 357-66
8.. Haroche J, Charlotte F, Arnaud L, High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses: Blood, 2012; 120(13); 2700-3
9.. Durham BH, Roos-Weil D, Baillou C, Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells: Blood, 2017; 130(2); 176-80
10.. Haroche J, Cohen-Aubart F, Arnaud L, [Erdheim-Chester disease.]: Rev Med Interne, 2014; 35; 715-22 [in French]
11.. Cangi MG, Biavasco R, Cavalli G, BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim-Chester disease: Ann Rheum Dis, 2015; 74(8); 1596-602
12.. Lachenal F, Cotton F, Desmurs-Clavel H, Neurological manifestations and neuroradiological presentation of Erdheim-Chester disease: Report of 6 cases and systematic review of the literature: J Neurol, 2006; 253(10); 1267-77
13.. Chasset F, Barete S, Charlotte F, Cutaneous manifestations of Erdheim-Chester disease (ECD): Clinical, pathological, and molecular features in a monocentric series of 40 patients: J Am Acad Dermatol, 2016; 74(3); 513-20
14.. Courtillot C, Laugier Robiolle S, Cohen Aubart F, Endocrine manifestations in a monocentric cohort of 64 patients with Erdheim-Chester disease: J Clin Endocrinol Metab, 2016; 101(1); 305-13
15.. Diamond EL, Dagna L, Hyman DM, Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease: Blood, 2014; 124(4); 483-92
16.. Pegoraro F, Papo M, Maniscalco V, Erdheim-Chester disease: A rapidly evolving disease model: Leukemia, 2020; 34; 2840-57
17.. Clements MB, Farhi J, Schenkman NS, Erdheim-Chester disease presenting as a solid renal mass: Urology, 2017; 100; e1-2
18.. Wu Z, Jiang G-L, Tang Y, Urinary involvement in Erdheim-Chester disease: Computed tomography imaging findings: Abdom Radiol (NY), 2021; 46(9); 4324-31
19.. Papo M, Diamond EL, Cohen-Aubart F, High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis: Blood, 2017; 130(8); 1007-13
20.. Haroche J, Cohen-Aubart F, Amoura Z, Erdheim-Chester disease: Blood, 2020; 135(16); 1311-18
21.. Couvreur T, Lipcsei G, Nchimi A, Imaging assessment of periaortic inflammation in Erdheim-Chester disease: Aorta (Stamford), 2013; 1(2); 146-48
22.. Rodríguez Sánchez D, Saura Espín D, Bas Bernal Á, Erdheim-Chester syndrome with periaortitis: Rev Esp Cardiol (Engl Ed), 2016; 69(1); 72
23.. Haroche J, Amoura Z, Dion E, Cardiovascular involvement, an overlooked feature of Erdheim-Chester disease: Report of 6 new cases and a literature review: Medicine (Baltimore), 2004; 83(6); 371-92
24.. Al Hinai J, Al Sibani M, Al-Maqbali JS, Al Alawi AM, Pericardial effusion in Erdheim-Chester disease: A case report and a mini literature review: Cureus, 2022; 14(2); e22010
25.. Lutz SZ, Schmalzing M, Vogel-Claussen J, Adam P, May AE, [Recurrent pericardial effusion as first manifestation of Erdheim-Chester disease.]: Dtsch Med Wochenschr, 2011; 136(39); 1952-56 [in German]
26.. Nakhleh A, Slobodin G, Elias N, Rapid progression to cardiac tamponade in Erdheim-Chester disease despite treatment with interferon alpha: Mod Rheumatol, 2016; 26(4); 621-24
27.. Ivan D, Neto A, Lemos L, Gupta A, Erdheim-Chester disease: A unique presentation with liver involvement and vertebral osteolytic lesions: Arch Pathol Lab Med, 2003; 127(8); e337-39
28.. Klieger MR, Schultz E, Elkowitz DE, Erdheim-Chester disease: A unique presentation with multiple osteolytic lesions of the spine and pelvis that spared the appendicular skeleton: Am J Roentgenol, 2002; 178(2); 429-32
29.. Chinchilla EA, Gourde M-P, Turcotte K, Case of Erdheim-Chester presenting with xanthelasma-like eruption and osteolytic bone lesions: A case report: SAGE Open Med Case Rep, 2019; 7; 2050313 X19845217
30.. Yalamanchi A, Asirvatham AR, Balachandran K, Erdheim-Chester disease – unusual presentation with isolated skeletal lytic lesions: J Orthop Case Rep, 2022; 12(1); 63-67
31.. Gupta A, Aman K, Al-Babtain M, Multisystem Erdheim-Chester disease; A unique presentation with liver and axial skeletal involvement: Br J Haematol, 2007; 138(3); 280
32.. Chistiakov DA, Killingsworth MC, Myasoedova VA, CD68/macrosialin: Not just a histochemical marker: Lab Invest, 2017; 97(1); 4-13
33.. Harmon CM, Brown N, Langerhans cell histiocytosis: A clinicopathologic review and molecular pathogenetic update: Arch Pathol Lab Med, 2015; 139(10); 1211-14
34.. Naeem M, Menias CO, Cail AJ, Imaging spectrum of granulomatous diseases of the abdomen and pelvis: Radiographics, 2021; 41(3); 783-801
35.. Hornick JL, Fletcher CDM, PEComa: What do we know so far?: Histopathology, 2006; 48(1); 75-82
Figures
In Press
03 Apr 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943392
04 Apr 2024 : Case report ")
Am J Case Rep In Press; DOI: 10.12659/AJCR.943271
05 Apr 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943506
05 Apr 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.942717
Most Viewed Current Articles
07 Mar 2024 : Case report
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133
10 Jan 2022 : Case report 
DOI :10.12659/AJCR.935263
Am J Case Rep 2022; 23:e935263
19 Jul 2022 : Case report 
DOI :10.12659/AJCR.936128
Am J Case Rep 2022; 23:e936128
23 Feb 2022 : Case report
DOI :10.12659/AJCR.935250
Am J Case Rep 2022; 23:e935250