26 March 2024: Articles 
A Rare Case of Neonatal Hypomagnesemia with Secondary Hypocalcemia Caused by a Novel Homozygous TRPM6 Gene Variant
Challenging differential diagnosis, Management of emergency care, Rare disease, Congenital defects / diseases
Mohammed Shahab UddinDOI: 10.12659/AJCR.942498
Am J Case Rep 2024; 25:e942498






Abstract
BACKGROUND: Familial hypomagnesemia with secondary hypocalcemia (HSH) is a rare autosomal recessive disorder (OMIM# 602014) caused by mutations in the gene encoding transient receptor potential melastatin 6 (TRPM6)) on chromosome 9q22, a channel involved in epithelial magnesium resorption. While a plethora of studies have delineated various clinical manifestations pertinent to this mutation, the literature is devoid of connections between TRPM6 mutations and bleeding diathesis, or sudden infant death syndrome (SIDS). This report presents a case of familial HSH associated with the novel homozygous TRPM6 gene variant c.5281C>G p. (Arg1761Gly) chr9: 77354845.
CASE REPORT: This report details a 26-day-old neonate, born full term with optimal Apgar scores, who experienced an abrupt emergence of apnea, cyanosis, bilateral nasal bleeding, and diminished alertness. Despite the neonate’s initially unremarkable clinical birth indicators, a meticulous assessment unveiled a pronounced family history of SIDS, including a sibling previously diagnosed with hypomagnesemia. Laboratory examination of the infant demonstrated severe hypomagnesemia and hypocalcemia, conditions which were promptly ameliorated following intravenous administration of magnesium and calcium. Whole-exome sequencing identified a homozygous TRPM6 gene mutation c.5281C>G p. (Arg1761Gly) at chr9: 77354845. This gene is crucial for magnesium regulation. The mutation involves a cytosine-to-guanine shift, resulting in an arginine to glycine amino acid substitution at position 1761 of the TRPM6 protein.
CONCLUSIONS: This report has highlighted that infantile hypomagnesemia may be associated with symptoms and signs that can mimic infection, or it can present with seizures. Although familial HSH is a rare genetic disorder that can be identified by genetic testing, correction of hypomagnesemia is the most important and immediate clinical management strategy.
Keywords: Hypomagnesemia 1, Intestinal, Neonatal sepsis, Sudden Infant Death, TRPM6 Protein, Human, Whole Exome Sequencing
Introduction
Magnesium is indispensable in the body, serving as a cofactor in over 300 enzymatic reactions encompassing energy production, DNA synthesis, and muscle and nerve function. Its significance extends to maintaining bone health and regulating cardiovascular function, underlining its pivotal role in overall physiological well-being [1,2]. Thus, a comprehensive understanding of magnesium absorption mechanisms is vital. This involves both passive paracellular transport and active transcellular transport, with the latter 2 processes regulated by 2 highly homologous magnesium channels, transient receptor potential melastatin channel types 6 and 7 (TRPM6 and TRPM7) [3–6]. These channels are predominantly located in the gastrointestinal tract and the distal convoluted tubules [7–9]. TRPM6 is specifically associated with epithelial magnesium transfer, while TRPM7 plays a crucial role in maintaining magnesium homeostasis [10–12]. Loss-of-function mutations in TRPM6 are linked to autosomal recessive familial hypomagnesemia with secondary hypocalcemia (HSH, OMIM 602014) [13–15]. Although renal magnesium wasting was initially believed to be the primary cause of low magnesium levels in HSH, recent studies suggest that TRPM6 expression defects, influencing intestinal magnesium uptake, may also play a pivotal role in regulating intestinal magnesium absorption [16]. Moreover, Chubanov et al (2004) highlighted the potential significance of TRPM6/TRPM7 hetero-oligomerization for TRPM6’s physiological function in epithelial magnesium absorption. The binding of TRPM6 and TRPM7 to form an ion channel complex in the cell membrane implies that mutations in the TRPM6 gene disrupt TRPM6/TRPM7 complex formation, leading to HSH [17]. Consequently, rare TRPM7 variants are also implicated in causing hypomagnesemia with secondary hypocalcemia (HSH) [16]. Additionally, the spectrum of genetic causes of neonatal hypomagnesemia is broad. Gitelman syndrome, arising from SLC12A3 mutations, disrupts magnesium reabsorption, resulting in hypomagnesemia and hypercalciuria [3]. Hypocalciuric hypomagnesemia, attributed to mutations in the thick ascending limb of Henle, leads to hypercalciuric hypomagnesemia [4]. Furthermore, epilepsy, ataxia, sensorineural deafness (EAST) syndrome, stemming from KCNJ10 mutations, culminates in hypomagnesemia due to impaired Kir4.1 potassium channel function [6,7]. Homozygous or compound heterozygous mutations in CLCNKB, encoding the chloride ion (Cl–)channel ClC-Kb, are responsible for Bartter syndrome type III. In addition, FXYD2 is associated with isolated dominant hypomagnesemia, and recessive mutations in PCBD1 also constitute genetic causes of hypomagnesemia [8–10].
The clinical presentation of TRPM6-related autosomal recessive familial HSH has been investigated, and seizure is the most common acute finding [18], often with neurological damage due to late diagnosis [18,19]. At a mean age of 4.9 (±2.5) weeks, Shalev et al (1998) described 15 cases of 60% hypocalcemic-hypomagnesemic seizures with permanent neurological damage [20]. Infants have presented with carpopedal spasms, hemiparesis, and aphasia [21,22]; basal ganglia calcification [23]; arrhythmia [24]; misdiagnosis as nutritional rickets [25]; repeated self-terminating Torsades de pointes [26]; and hypertensive urgency with unknown mechanisms [27]. The potential relevance of infantile apnea and sudden infant death syndrome (SIDS) caused by severe hypomagnesemia has also been considered [28]. To the best of our knowledge, no case reports of bleeding diathesis or near-miss SIDS have been reported in association with the TRPM6 gene mutation.
A 26-day-old neonate presented in our clinic with sepsis-like symptoms and a catastrophic life-threatening event caused by severe hypomagnesemia with hypocalcemia with bleeding tendency. There was also a family history of SIDS. Whole-exome sequencing revealed a mutation in the TRPM6 gene c.5281C>G p. (Arg1761Gly) chr9: 77354845 that had not been known before.
Case Report
WHOLE-EXOME SEQUENCING METHODOLOGY:
Genomic DNA was purified and fragmented, with subsequent enrichment and amplification of the exons of all recognized human genes and associated exon-intron boundaries utilizing the Roche NimbleGen capture methodology (SeqCap MedExome Library). Sequencing was performed through Illumina technology, ensuring an average coverage depth of 80-fold. Approximately 98% and 96% of targeted regions achieved coverage depths of 15-fold and 20-fold, respectively. The gathered data were mapped to the hg19 genome assembly, with variant identification and annotation executed using a proprietary bioinformatics pipeline (Bioscientia, Ingelheim, Germany). Detected single nucleotide variants and insertion-deletions (indels) underwent filtering against both internal and external databases, emphasizing rare variants (Genome Aggregation Database [gnomAD] minor allele frequency ≤1%) and omitting recognized artifacts or variants from regions of high homology. Variant classification adhered to the American College of Medical Genetics guidelines, incorporating database entries, bioinformatics prediction tools, and current literature. Potentially pathogenic variations contrasting the wild-type sequence (hg19, GRCh37) and the patient’s sequence were detailed in this report after rigorous quality assessment. Quality-compromised variants underwent confirmation via PCR amplification and Sanger sequencing. Sample identity assurance was embedded in the quality management protocol.
The WES results brought to light a homozygous variation in the TRPM6 gene: c.5281C>G p. (Arg1761Gly) chr9: 77354845 (Figure 2). The TRPM6 gene, positioned on chromosome 9 at locus 77354845, is integral to magnesium homeostasis. A point mutation was identified at position 5281 within this gene; a substitution point mutation of cytosine (C) to guanine (G). This nucleotide substitution mutation resulted in a consequential amino acid substitution from arginine to glycine at residue 1761 of the encoded TRPM6 protein. Remarkably, the neonate exhibited homozygosity for this variant, implying that both allelic copies of the TRPM6 gene bore the identical mutation. Such a homozygous state could manifest a more accentuated phenotype relative to heterozygous counterparts. Arginine, with its pronounced positive charge and substantial structure, contrasts starkly with the small, neutral glycine. This stark chemical divergence might engender functional perturbations in the TRPM6 protein, potentially undermining its role in magnesium regulation. The identified variant, Arg1761Gly, had not been previously documented in the available literature or databases at the time of this report. In silico analysis using multiple algorithms (including PolyPhen-2, SIFT, and MutationTaster) predicted this variation to be deleterious, implying a potential association with the neonate’s clinical phenotype. On consultation with metabolic genetics, it was surmised that the neonate’s profound hypomagnesemia and secondary hypocalcemia could be attributed to a dysfunctional TRPM6 channel, leading to impaired intestinal absorption and renal reabsorption of magnesium. This finding, coupled with a similar clinical picture in the neonate’s sibling, indicated an autosomal recessive inheritance pattern. A retrospective evaluation of the sibling’s medical records corroborated the clinical phenotype and the TRPM6 gene variant, further bolstering the diagnosis. The family was counseled regarding the implications of this genetic alteration, emphasizing its autosomal recessive inheritance pattern and the risk to future offspring.
Discussion
The WES results brought to light a homozygous variation in the TRPM6 gene: c.5281C>G p. (Arg1761Gly) chr9: 77354845.
The described homozygous variation in the TRPM6 gene, denoted as c.5281C>G p. (Arg1761Gly) located on chromosome 9 at the position marked 77354845, indicates a precise genetic alteration whereby our patient inherited 2 identical mutations, 1 from each parent. The notation “c.5281C>G” specifies a substitution at the nucleotide level whereby the cytosine (C) was replaced by guanine (G) at the 5281st base pair in the coding sequence of the gene. This single nucleotide polymorphism results in a corresponding amino acid change at the protein level, described as p. (Arg1761Gly), whereby the normal arginine at position 1761 is replaced by glycine, potentially altering the protein’s function. The location “chr9: 77354845” pinpoints this mutation to the ninth chromosome, providing a specific genomic address that facilitates precise identification and study of this variant’s impact on health. TRPM6 is essential for the intestinal absorption of magnesium. TRPM6 gene mutations have been linked to hereditary HSH, a rare inherited condition [10,12]. This condition manifests in newborns or young infants, with symptoms including restlessness, shaking, muscle cramps, twitching, bluish skin around the mouth, and seizures [20,29]. The root cause of these symptoms is a problem with magnesium absorption in the intestines, which results in extremely low magnesium levels in the body. Furthermore, magnesium deficiency lowers calcium levels, primarily by interfering with the production and release of parathyroid hormone [12,30]. Current research, however, does not provide much information about other potential symptoms such as bleeding and conditions that mimic sepsis, as well as SIDS.
Magnesium is essential for blood clotting. It is crucial for the activation of one clotting component, factor X, by another, factor IXa. Other elements, such as factor VIIIa, phospholipids, and calcium ions, enhance this activation [31]. As a result, previous research may have overlooked the role of factors IX and VIII in this process. Animal studies suggest that when magnesium levels are extremely low, abnormal platelet activation can increase the risk of death [28]. Magnesium is required for proper blood clotting, platelet shape maintenance, and their ability to stick together [31–33]. Magnesium, as the fourth most common ion inside cells, is undeniably important for many bodily functions, including clotting [34,35]. When magnesium levels are low, the body’s ability to properly clot blood is compromised. This can have an impact on the activation of clotting factors, fibrin formation, and the risk of bleeding [36,37]. Furthermore, low magnesium levels can impair platelet function by affecting integrin and calcium balance. This reduces the ability of platelets to stick together, increasing the likelihood of bleeding [38]. Given this, we suspect that the reported nosebleed in our case was caused by a severe magnesium deficiency. It suggests that we should investigate magnesium’s role in blood clotting further, particularly when linked to HSH caused by TRPM6 mutations.
JL Caddell (2009) proposed that a magnesium deficiency relative to calcium in stressed infants could contribute to the onset of SIDS [39,40]. This disparity may be more pronounced in neonates from low-income families living in high-stress urban areas. Furthermore, preterm infants may be at greater risk due to decreased magnesium reserves caused by certain medical treatments. This could cause platelet aggregation and the release of thromboxane A2 (TXA2), potentially resulting in apnea and death. Although determining magnesium deficiency postmortem is difficult, research on magnesium supplementation may shed light on this possibility. James K Miller (1990) investigated the effects of hypomagnesemia on platelet responsiveness in bovine and ovine models, implying a possible link between infantile apnea and SIDS [28]. TRPM6 gene mutation-related severe hypomagnesemia could explain cases of SIDS, especially if a sibling has a family history. Over the last few decades, there has been a significant evolution in our understanding and characterization of acute, unexplained events in infants. Historically, the term apparent life-threatening event (ALTE) was used to describe incidents in infants that caused significant concern because of symptoms such as apnea, altered skin tone, drastic changes in muscular tone, choking, or gagging [41]. However, this definition raised concerns about its possible overlap with the ominous SIDS. Concurrent hypocalcemia and hypomagnesemia have been identified as causative agents in a subset of ALTE episodes, potentially inducing these events via epileptogenic activities or other yet-to-be-determined pathways [42]. The repercussions of magnesium deficiency are severe, with substantial evidence highlighting its ability to cause lethal cardiac arrhythmias, increasing the risk of sudden infant death [39,42–44]. In the present case, the index patient had a sibling who succumbed to SIDS at 3 months of age in the emergency department, underscoring the potential hereditary implications of a mutated TRPM6 gene within this family.
Historically, ALTE cases that required cardiopulmonary resuscitation were frequently referred to as “near-miss SIDS” [45]. When applied to our current case, where resuscitation measures were required, the question arises: could it be precisely referred to as a “near-miss SIDS” event? Because of the severity of our case’s clinical presentation, we must classify it as ALTE or near-miss SIDS. However, the discovery of a homozygous TRPM6 gene variant in our patient, which resulted in severe hypomagnesemia, provides a concrete etiological framework that distinguishes it from the idiopathic nature often associated with SIDS cases. The link between the identified severe hypomagnesemia caused by the TRPM6 gene variant and the infant’s clinical manifestation emphasizes the importance of thorough diagnostic efforts in such cases. It is critical to distinguish between truly unexplained episodes and those with identifiable etiologies, as this influences therapeutic strategies, prognosis, and familial guidance. Furthermore, diagnosis of neonatal sepsis is challenging because the symptoms in newborns can be varied and unclear [46]. Previous research has shown that newborns with sepsis can exhibit a wide range of symptoms. Unstable vital signs, feeding issues, changes in brain function, and abnormal muscle tone are all examples [47]. Because sepsis is such a serious threat, a quick and accurate diagnosis is critical to begin life-saving treatments as soon as possible [48]. In the present case, the clinical presentation included symptoms consistent with both neonatal sepsis and severe HSH, ultimately attributed to a TRPM6 gene mutation (c.5281C>G, p. Arg1761Gly). The clinical manifestations of hypomagnesemia, which may include neuromuscular irritability, tremors, and convulsions, closely mimic those of neonatal sepsis [49], thus complicating the diagnostic process. Consequently, the patient was administered a 72-hour course of antibiotics until cultures from blood, urine, and cerebrospinal fluid were confirmed to be negative.
Neonatal hypomagnesemia is frequently caused by genetic abnormalities that alter magnesium intake and regulation. TRPM6 stands out as a crucial gene for magnesium absorption in the intestines and kidneys, with mutations causing autosomal recessive hypomagnesemia and subsequent hypocalcemia, as we discovered in our case. Similarly, the Claudin-16 (CLDN16) and Claudin-19 (CLDN19) genes have been linked to familial hypomagnesemia with hypercalciuria and nephrocalcinosis, both of which produce significant magnesium deficiency [25]. The FXYD domain-containing ion transport regulator 2 (FXYD2) gene codes for the gamma subunit of the Na+/K+-ATPase pump, and mutations in this gene have been related to isolated dominant hypomagnesemia [50]. Mutations of another important gene, Hepatocyte Nuclear Factor 1 Homeobox B (HNF1B), cause renal magnesium wasting [51]. Furthermore, mutations of 4 other genes have been linked to hypomagnesemia: (1) Solute Carrier Family 12 Member 3 (SLC12A3), which encodes a thiazide-sensitive sodium-chloride transporter [52]; (2) Bartter syndrome, infantile, with sensorineural deafness (BSND), which is linked to Bartter syndrome and hypomagnesemia [53]; (3) Potassium Inwardly Rectifying Channel Subfamily J Member 10 (KCNJ10), which is linked to EAST syndrome [54]; and (4) Cyclin and CBS-Domain Divalent Metal Cation Transport Mediator 2 (CNNM2), which is linked to renal and intestinal magnesium wasting [55]. All 4 of these genes are integral in a variety of magnesium-related pathways. Mutations of these genes play important roles in genetic causes of hypomagnesemia, and can culminate in the clinical manifestation of newborn hypomagnesemia, highlighting the complexities in our case described in this report.
HSH, commonly presenting within the first 3 months of life, is a genetic condition often stemming from TRPM6 gene mutations. The management of this disorder necessitates a meticulous, individualized approach. High-dose oral magnesium supplementation remains the cornerstone of treatment, with initial dosing typically at 0.6 mmol/kg/day, though this can vary from 0.41 to 3.90 mmol/kg/day [19]. While magnesium oxide, magnesium chloride, or magnesium sulfate may be used, magnesium chloride or magnesium lactate formulations have been associated with reduced incidence of loose stools. Management is not limited to magnesium supplementation. Adjunctive therapies include calcium supplementation in the presence of symptomatic hypocalcemia, and in refractory cases, medications like amiloride can be used to minimize renal magnesium wasting. Vitamin D and its analogs may be beneficial for enhancing the intestinal absorption of calcium and magnesium. Some clinical improvement has been linked to spironolactone, which may elevate serum magnesium [48], and supportive measures such as dietary adjustments, probiotic administration, and improved familial care have also shown positive effects. The complexity of HSH management underscores the necessity for a multidisciplinary team approach, incorporating the expertise of neonatologists, pediatric nephrologists, dietitians, and genetic counselors. This collaborative strategy not only addresses the immediate and long-term clinical needs but also facilitates genetic counseling for families, given the autosomal recessive inheritance pattern of TRPM6 mutations. In fact, the nuanced management of neonatal hypomagnesemia due to TRPM6 gene mutations requires a delicate balance between immediate intervention and long-term care strategies. Through personalized treatment plans and a multidisciplinary team approach, the prognosis of HSH patients can be significantly improved, ensuring a comprehensive care paradigm that spans from infancy potentially into adulthood.
Familial HSH is linked to a spectrum of TRPM6 gene mutations, each uniquely altering the protein’s structure and magnesium transport function. The c.3449G>A (p.Arg1150Gln) mutation disrupts amino acid interactions, potentially affecting the channel structure and magnesium transportation [56]. Two other mutations, c.4934C>T (p.Ala1645Val) and c.2218C>T (p.Thr740Met), involve substitutions that might alter the protein’s tertiary structure or interaction with magnesium [57]. The c.1756G>A (p.Gly586Arg) mutation introduces a charge distribution change, affecting the protein’s configuration [58]. Particularly impactful are c.3952delC (p.Leu1318fs) and c.3629–3631delAAT (p.Met1210del) mutations, which can drastically alter the protein’s structure through frameshift or amino acid deletion, leading to significant functional disruptions [59]. Similarly, c.2722C>T (p.Pro908Ser) involves a substitution that could change the local protein structure [11]. These mutations collectively elucidate the pathophysiology of familial HSH, highlighting the critical role of the TRPM6 protein in maintaining magnesium balance and the diverse genetic underpinnings that lead to the condition. This genetic diversity has been expanded by the newly found c.5281C>G p. (Arg1761Gly) mutation in our case. Discovery of this mutation in our patient expands our understanding of the relationship between genotype and phenotype and the mechanics of magnesium-related disease. The new TRPM6 homozygous variant c.5281C>G p. (Arg1761Gly) plays a critical role in magnesium control and may modify the structure, trafficking, and kinase activity of the TRPM6 protein. Protein stability, ion conductance, and channel stability can all be affected by glycine replacing arginine. It may also prevent the ion channel, which is essential for magnesium transport, from being expressed on the surface of cells and from being properly localized. Potentially affecting magnesium homeostasis, the mutation may alter the kinase domain’s functionality, which in turn may affect regulatory phosphorylation and protein-protein interactions. Investigating these pathways further is crucial for understanding the pathophysiology of diseases like secondary hypocalcemia and familial hypomagnesemia as well as for identifying possible treatment options.
In addition, the finding of the new TRPM6 gene variant c.5281C>G p. (Arg1761Gly) chr9: 77354845 in a family that has a history of SIDS, hypomagnesemia, and a near-SIDS occurrence strongly suggests that this mutation may have far-reaching repercussions. The Arg1761Gly substitution may cause more than just the known phenotype of HSH; it may also affect life-threatening conditions like SIDS, because TRPM6 is involved in magnesium homeostasis. This mutation may impact multiple biological processes, since it has been detected in this family but has not been associated with sepsis or bleeding in previous research. To determine its precise effects on kinase function, protein location, and ion channel functioning, further research is required. The severity of the mutation and its consequences for future generations make genetic counseling an absolute necessity for this family. To fully understand how the Arg1761Gly mutation impacts TRPM6 function and contributes to the diverse clinical phenotypes observed, it is essential that researchers work together, especially through functional assays in model organisms or cell lines. This will allow for the development of targeted interventions and management strategies.
Conclusions
In conclusion, this case report adds to the growing body of evidence that genetic mutations affecting electrolyte balance can present life-threatening symptoms in neonates and infants. It calls for a broadened scope of consideration beyond common diagnoses to encompass rare genetic mutations, such as those in the TRPM6 gene, which may manifest with severe clinical consequences. Furthermore, it serves as a testament to the evolution of pediatric care, where the integration of advanced genetic insights into clinical practice is becoming increasingly vital for timely and accurate diagnoses, ensuring the best possible outcomes for patients.
Figures
References:
1.. de Baaij JHF, Hoenderop JGJ, Bindels RJM, Magnesium in man: Implications for health and disease: Physiol Rev, 2015; 95(1); 1-46
2.. Saris NEL, Mervaala E, Karppanen H, Magnesium: An update on physiological, clinical and analytical aspects: Clin Chim Acta, 2000; 294(1–2); 1-26
3.. Luongo F, Pietropaolo G, Gautier M, TRPM6 is essential for magnesium uptake and epithelial cell function in the colon: Nutrients, 2018; 10(6); 784
4.. Vargas-Poussou R, Claverie-Martin F, Prot-Bertoye C, Possible role for rare TRPM7 variants in patients with hypomagnesemia with secondary hypocalcemia: Nephrol Dial Transplant, 2022; 38(3); 1-12
5.. Pietropaolo G, Pugliese D, Armuzzi A, Magnesium absorption in intestinal cells: Evidence of cross-talk between EGF and TRPM6 and novel implications for cetuximab therapy: Nutrients, 2020; 12(11); 3277
6.. Bindels RJM, Calciotropic and magnesiotropic TRP channels: Physiology, 2008; 23(1); 32-40
7.. Cao G, Lee K, Van Der Wijst J, Methionine sulfoxide reductase B1 (MsrB1) recovers TRPM6 channel activity during oxidative stress: J Biol Chem, 2010; 285(34); 26081-87
8.. Groenestege WMT, Rayssiguier Y, Mazur A, Relationship between low magnesium status and TRPM6 expression in the kidney and large intestine: Am J Physiol Regul Integr Comp Physiol, 2008; 294(6); R2001-R7
9.. Voets T, Nilius B, Hoefs S, TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption: J Biol Chem, 2004; 279(1); 19-25
10.. Chubanov V, Gudermann ÆT, Schlingmann KP, Essential role for TRPM6 in epithelial magnesium transport and body magnesium homeostasis: Pflugers Arch, 2005; 451(1); 228-34
11.. Schlingmann KP, Waldegger S, Konrad M, TRPM6 and TRPM7 – Gatekeepers of human magnesium metabolism: Biochim Biophys Acta, 2007; 1772(8); 813-21
12.. Schlingmann KP, Gudermann T, A critical role of TRPM channel-kinase for human magnesium transport: J Physiol, 2005; 566(Pt 2); 301-8
13.. Apa H, Kayserili E, Agin H, A case of hypomagnesemia with secondary hypocalcemia caused by TRPM6 gene mutation: Indian J Pediatr, 2008; 75(6); 632-34
14.. Patel S, Rayanagoudar G, Gelding S, Familial hypomagnesaemia with secondary hypocalcaemia: BMJ Case Rep, 2016; 2016; bcr2016216870
15.. Schlingmann KP, Weber S, Peters M, Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family: Nat Genet, 2002; 31(2); 166-70
16.. Landau D, Meyer P, Shalev H, Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia: Nat Genet, 2002; 31(2); 171-74
17.. Pham PC, Pham PA, Pham SV, Hypomagnesemia: A clinical perspective: Int J Nephrol Renovasc Dis, 2014; 7; 219-30
18.. Schlingmann KP, Sassen MC, Weber S, Novel TRPM6 mutations in 21 families with primary hypomagnesemia and secondary hypocalcemia: J Am Soc Nephrol, 2005; 16(10); 3061-69
19.. Katayama K, Povalko N, Yatsuga S, Nishioka J, New TRPM6 mutation and management of hypomagnesaemia with secondary hypocalcaemia: Brain Dev, 2015; 37(3); 292-98
20.. Shalev H, Phillip M, Galil A, Clinical presentation and outcome in primary familial hypomagnesaemia: Arch Dis Child, 1998; 78(2); 127-30
21.. Dharnidharka VR, Carney PR, Hypomagnesemia presenting as aphasia: Pediatr Neurol, 2005; 33(1); 61-65
22.. Visudhiphan P, Visudtibhan A, Chiemchanya S, Khongkhatithum C, Neonatal seizures and familial hypomagnesemia with secondary hypocalcemia: Pediatr Neurol, 2005; 33(3); 202-5
23.. Habeb AM, Al-Harbi H, Schlingmann KP, Resolving basal ganglia calcification in hereditary hypomagnesemia with secondary hypocalcemia due to a novel TRPM6 gene mutation: Saudi J Kidney Dis Transpl, 2012; 23(5); 1038-42
24.. Dimke H, Hoenderop JGJ, Bindels JM, Molecular basis of epithelial Ca2+ and Mg2+ transport: Insights from the TRP channel family: J Physiol, 2011; 589(Pt 7); 1535-42
25.. Kari JA, Farouq M, Alshaya HO, Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: Pediatr Nephrol, 2003; 18(6); 506-10
26.. Mahadevappa M, Cardiologist C, Severe hypomagnesemia with secondary hypocalcaemia presenting as recurrent self-terminating Torsades de pointes: IHJ Cardiovasc Case Rep, 2020; 4(3); 138-41
27.. Satyajit B, Prasad VV, Hypomagnesemia presenting as hypertensive urgency and multiple focal seizures in an infant: J Pediatr Crit Care, 2019; 6(4); 37
28.. Miller JK, Schneider MD, Ramsey N, Effects of hypomagnesemia on reactivity of bovine and ovine platelets: Possible relevance to infantile apnea and sudden infant death syndrome: J Am Coll Nutr, 1990; 9(1); 58-64
29.. Guran T, Akcay T, Bereket A, Clinical and molecular characterization of Turkish patients with familial hypomagnesaemia: Novel mutations in TRPM6 and CLDN16 genes: Nephrol Dial Transplant, 2012; 27(2); 667-73
30.. Srinivasan R, Catherine A, Sushma N, Anil K, Familial hypomagnesemia with hypocalcemia: A rare cause of infantile seizures: CEN Case Rep, 2022; 12(2); 139-45
31.. Sekiya F, Yoshida M, Yamashita T, Morita T, Magnesium (II) is a crucial constituent of the blood: Potentiation of coagulant activities of: J Biol Chem, 1996; 271(15); 8541-44
32.. Jankun J, Skrzypczak-Jankun E, Lipinski B, Complex function of magnesium in blood clot formation and lysis: Cent Eur J Immunol, 2013; 38(2); 149-53
33.. Des Prez RM, Bryant RE, Katz JA, Brittingham TE, Platelet aggregation by magnesium ion: Thromb Diath Haemorrh, 1967; 17(3–4); 516-31
34.. Kresge N, Simoni RD, Hill RL, The waterfall sequence for blood clotting: The work of Earl W: Davie. J Biol Chem, 2006; 281(48); e39
35.. Varga-Szabo D, Pleines I, Nieswandt B, Cell adhesion mechanisms in platelets: Arterioscler Thromb Vasc Biol, 2008; 28(3); 403-13
36.. Ames WA, McDonnell N, Potter D, The effect of ionised magnesium on coagulation using thromboelastography: Anaesthesia, 1999; 54(10); 999-1001
37.. Tangvoraphonkchai K, Davenport A, Magnesium and cardiovascular disease: Adv Chronic Kidney Dis, 2018; 25(3); 251-60
38.. Sheu JR, Hsiao G, Shen MY, Mechanisms involved in the antiplatelet activity of magnesium in human platelets: Br J Haematol, 2002; 119(4); 1033-41
39.. Caddell JL, Magnesium deficiency in the cause of the sudden infant death syndrome: An updated hypothesis: Pediatr Asthma Allergy Immunol, 1991; 5(3); 191-226
40.. Erickson MM, Poklis A, Gantner GE, Tissue mineral levels in victims of sudden infant death syndrome I: Toxic metals-lead and cadmium: Pediatr Res, 1983; 17(10); 779-84
41.. Fu LY, Apparent life-threatening events: an update: Pediatr Emerg Care, 2012; 33(8); 361-69
42.. Bashir H, Crom D, Metzger M, Cisplatin-induced hypomagnesemia and cardiac dysrhythmia: Pediatr Blood Cancer, 2007; 49(6); 867-69
43.. Kadivar M, Shahbaznejad L, A neonate with multiple causes of apparent life-threatening event (ALTE): A case report: Iran J Neonatol, 2016; 7(3); 33-35
44.. Hansen BA, Bruserud Ø, Hypomagnesemia in critically ill patients: J Intensive Care, 2018; 6; 1
45.. Gleeson M, Clancy RL, Cox AJ, Mucosal immune responses to infections in infants with acute life-threatening events classified as “near-miss” sudden infant death syndrome: FEMS Immunol Med Microbiol, 2004; 42(1); 105-18
46.. Polin RA, Papile LA, Baley JE, Management of neonates with suspected or proven early-onset bacterial sepsis: Pediatrics, 2012; 129(5); 1006-15
47.. Kim F, Polin RA, Hooven TA, Neonatal sepsis: BMJ, 2020; 371; m3672
48.. Wolff ASB, Nedrebø B, Bratland E, Hypomagnesemia and functional hypoparathyroidism due to novel mutations in the Mg-channel TRPM6: Endocr Connect, 2015; 4(4); 215-22
49.. Gazzaz N, Alghamdi M, Familial hypomagnesemia with secondary hypocalcemia: A case report, 2021; 13(11); 11-14
50.. De Baaij JHF, Dorresteijn EM, Hennekam EAM, Recurrent FXYD2 p.Gly41Arg mutation in patients with isolated dominant hypomagnesaemia: Nephrol Dial Transplant, 2015; 30(6); 952-57
51.. Adalat S, Woolf AS, Johnstone KA, HNF1B mutations associate with hypomagnesemia and renal magnesium wasting: J Am Soc Nephrol, 2009; 20(5); 1123-31
52.. Riancho JA, Saro G, Sañudo C, Gitelman syndrome: Genetic and expression analysis of the thiazide-sensitive sodium-chloride transporter in blood cells: Nephrol Dial Transplant, 2006; 21(1); 217-20
53.. Viering DHHM, de Baaij JHF, Walsh SB, Genetic causes of hypomagnesemia, a clinical overview: Pediatr Nephrol, 2017; 32(7); 1123-35
54.. Bandulik S, Schmidt K, Bockenhauer D, The salt-wasting phenotype of EAST syndrome, a disease with multifaceted symptoms linked to the KCNJ10 K+ channel: Pflugers Arch, 2011; 461; 423-35
55.. Konrad M, Schlingmann KP, Inherited disorders of renal hypomagnesaemia: Nephrol Dial Transplant, 2014; 29(Suppl. 4); iv63-iv71
56.. Walder RY, Landau D, Meyer P, Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia: Nat Genet, 2002; 31(2); 171-74
57.. Schlingmann KP, Weber S, Peters M, Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family: Nat Genet, 2002; 31(2); 166-70
58.. Groenestege WMT, Thébault S, Van Der Wijst J, Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia: J Clin Invest, 2007; 117(8); 2260-67
59.. Schlingmann KP, Sassen MC, Weber S, Novel TRPM6 mutations in 21 families with primary hypomagnesemia and secondary hypocalcemia: J Am Soc Nephrol, 2005; 16(10); 3061-69
Figures
Tables
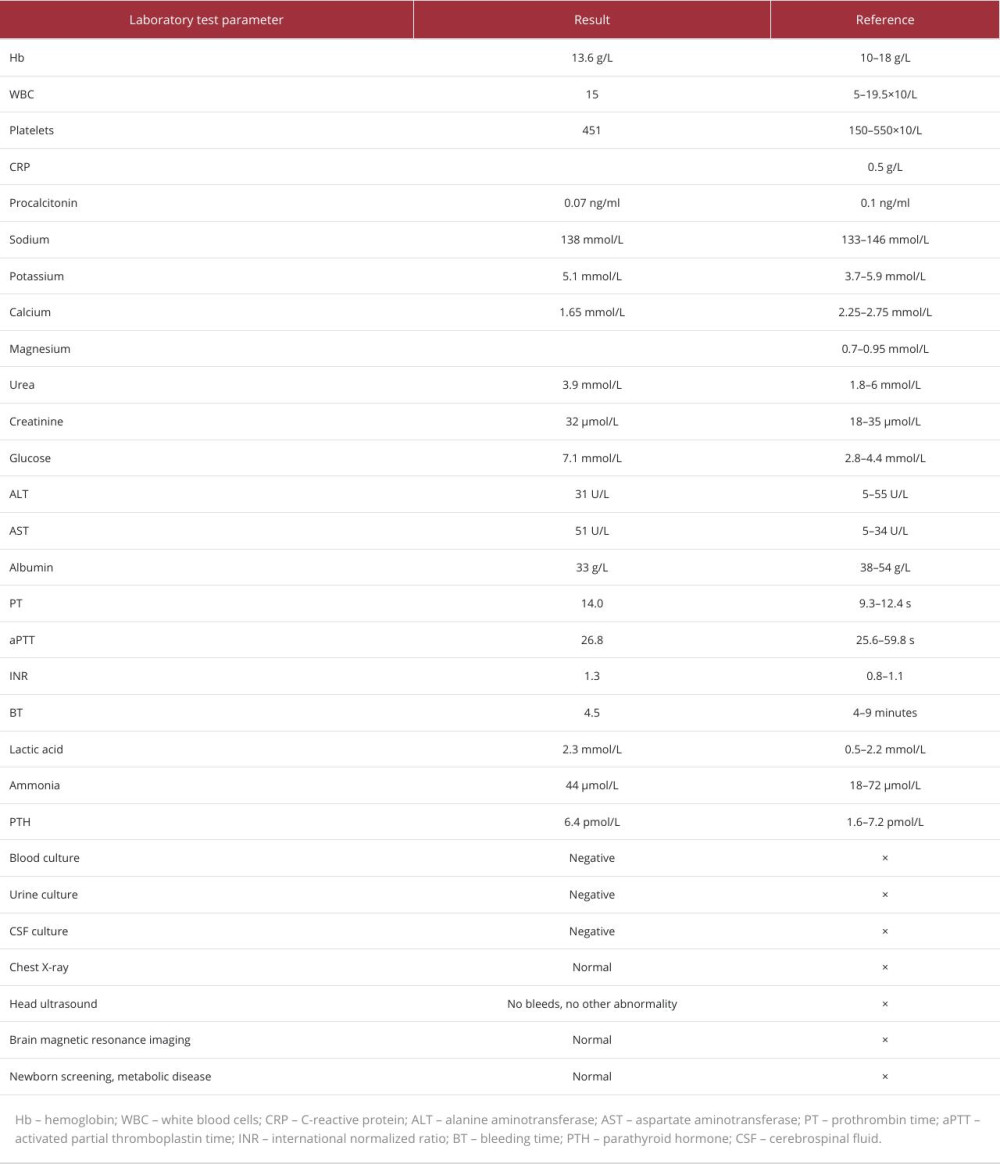 Table 1.. Comprehensive laboratory parameters obtained upon the patient’s admission. Values are presented alongside their respective reference intervals to highlight any clinically pertinent anomalies. Notably, deviations from the norm may signal underlying pathophysiological processes.Table 1.. Comprehensive laboratory parameters obtained upon the patient’s admission. Values are presented alongside their respective reference intervals to highlight any clinically pertinent anomalies. Notably, deviations from the norm may signal underlying pathophysiological processes.
Table 1.. Comprehensive laboratory parameters obtained upon the patient’s admission. Values are presented alongside their respective reference intervals to highlight any clinically pertinent anomalies. Notably, deviations from the norm may signal underlying pathophysiological processes.Table 1.. Comprehensive laboratory parameters obtained upon the patient’s admission. Values are presented alongside their respective reference intervals to highlight any clinically pertinent anomalies. Notably, deviations from the norm may signal underlying pathophysiological processes. In Press
19 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.942660
19 Mar 2024 : Case report ")
Am J Case Rep In Press; DOI: 10.12659/AJCR.943174
19 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943136
21 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943645
Most Viewed Current Articles
07 Mar 2024 : Case report 
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133
10 Jan 2022 : Case report 
DOI :10.12659/AJCR.935263
Am J Case Rep 2022; 23:e935263
19 Jul 2022 : Case report
DOI :10.12659/AJCR.936128
Am J Case Rep 2022; 23:e936128
23 Feb 2022 : Case report
DOI :10.12659/AJCR.935250
Am J Case Rep 2022; 23:e935250