18 July 2022: Articles 
A Case of a Rare Parathyroid Hormone (PTH)-Producing Neuroendocrine Tumor
Challenging differential diagnosis, Rare disease
Catherine Dianne Quinn1ABCDEFG*, Fihr Chaudhary2ABEF, Aron Gould-Simon3BD, Baorong Chen4BD, Harsimran Singh Bhandal5BEF, Uzair Chaudhary1ABDEGDOI: 10.12659/AJCR.935783
Am J Case Rep 2022; 23:e935783






Abstract
BACKGROUND: Neuroendocrine neoplasms are commonly seen in association with hormone production, and clinical signs that arise from these hormonal effects often manifest as the first presentation of malignancy. The excess production of parathyroid hormone (PTH) in particular, however, is primarily sporadic (80-85%) in clinical settings. In the context of malignancy, hyperparathyroidism manifestations arise most frequently from non-neuroendocrine pulmonary tumors through a ligand mimicker, parathyroid hormone-related peptide (PHrP). Excess PTH or PTHrP production has been very rarely described in association with gastrointestinal tumors and almost never described as a primary paraneoplastic syndrome from a neuroendocrine tumor (NET) alone.
CASE REPORT: We present a patient with a prior surgically resected carcinoid tumor who later presented with an elevated parathyroid hormone level, hypercalcemia, and clinical manifestations of primary hyperparathyroidism. She was found to have a low-grade, recurrent neuroendocrine tumor on resection of a parathyroid mass suspected to be a productive adenoma. Despite no longer having parathyroid glands given the extent of resection, her PTH level remained elevated and was rising. Further investigation via repeat sestamibi nuclear scan excluded the possibility of exogenous parathyroid tissue, and subsequent dotatate positron emission tomography/computed tomography (PET/CT) revealed the source of the PTH production: multiple sites of metastatic neuroendocrine tumors producing native PTH.
CONCLUSIONS: This case highlights the rare possibility of NETs to secrete PTH and the importance of considering early staging with dotatate PET/CT to evaluate the extent of disease. Additionally, our case reveals the importance of considering NET as an alternative etiology for refractory hypercalcemia.
Keywords: Parathyroid Hormone-Related Protein (1-108), neuroendocrine tumors, Paraneoplastic Endocrine Syndromes, Parathyroid Hormone, Gallium Ga 68 Dotatate, Positron Emission Tomography Computed Tomography, octreotide, Female, Humans, Hypercalcemia, Parathyroid Hormone-Related Protein, Positron-Emission Tomography, Radionuclide Imaging
Background
Primary hyperparathyroidism (PHPT) is caused by the excess secretion of parathyroid hormone (PTH) in the blood arising from hyperactive parathyroid glands. It is an inherited disorder in approximately 5% to 10% of all cases, but otherwise primary hyperparathyroidism tends to be acquired sporadically [1]. In these sporadic cases, 80% to 85% of them involve a non-malignant adenoma that produces high levels of parathyroid hormone resulting in hypercalcemia [2].
Hypercalcemia of malignancy is most often due to humoral mechanisms in malignancies that have metastasized to the bones or those that are productive of exogenous hormones, leading to paraneoplastic syndromes [2–4]. Most often, tumor cells produce a parathyroid-like protein known as parathyroid hormone-related peptide (PTHrP), which acts like PTH, causing increased serum calcium [2–4]. PTHrP and PTH bind to the same receptors in target tissues leading to downstream effects, including the rise in endogenous calcium levels [4]. Due to homeostatic feedback mechanisms, PTH levels will be low in patients with elevated PTHrP because calcium levels rise pathologically above normal ranges [4]. PTHrP and PTH associated with neuroendocrine tumors have been rarely described in the literature. PTH-producing tumors of gastrointestinal origin have been described in only 10 case reports, with only 4 cases having a neuroendocrine origin.
We report a case of a patient who developed malignancy-associated hypercalcemia in the setting of a neuroendocrine tumor due to the ectopic production of PTH.
Case Report
A 76-year-old woman with a past medical history of a grade 1 mesenteric carcinoid tumor of the small bowel, incidentally found during a small bowel resection in May 2009, was referred to our clinic in December 2019 after the following clinical course indicated recurrence of her neuroendocrine tumor with manifestations of a PTH-producing paraneoplastic syndrome.
The carcinoid tumor resected in 2009 had been found incidentally on pathology results during a small bowel resection for bowel necrosis during a traumatic event that the patient sustained. The pathology results demonstrated submucosal nodules consistent with carcinoid histology, situated 16 cm from the mucosal margin of the resection, ranging from 0.7 to 1.0 cm in size. The tumor was grade 1 in mitotic activity and well-differentiated. It extended through the muscularis propria, involving the serosa and mesentery, and coalesced into a 3.0 cm sclerotic mass. Thirteen lymph nodes evaluated during the original re-section were negative for evidence of carcinoid histology. The patient had been asymptomatic for neuroendocrine manifestations, and given that the tumor was low grade, she did not receive any adjuvant treatment thereafter.
In July 2019, she was found to have mildly elevated calcium levels (10.8 mg/dL) during a routine primary care evaluation. On further laboratory investigation, PTH was also noted to be elevated (100.7 pg/mL). In August 2019, the patient underwent a technetium 99 SPECT nuclear parathyroid scan, which demonstrated an approximate 1.5 cm mass, suspected to be a parathyroid adenoma, at the level of the right paratracheal space in the mediastinum.
During this time, the patient reported progressive diarrhea and flushing for several months as well as weight loss of 5.4 kg. She denied experiencing these symptoms during the discovery of her original carcinoid tumor in 2009. Although her calcium level was mildly elevated (10.8 mg/dL), the patient had normal vitamin D levels (1,25 dihydroxycholecalciferol) of 38 pg/mL. The level of serum phosphorus was also within the reference range (3.4 mg/dL). Despite the mild elevation, the patient described clinical manifestations of hypercalcemia as well. She endorsed increasing fatigue, bone pain in her left pelvis, left foot, and spinal column, palpitations lasting for several hours, new mood disturbances with anxiety, and worsening memory deficits that had arisen only in the prior few months. She denied abdominal discomfort, kidney stones, polydipsia, polyuria, nausea and vomiting, muscle weakness, and muscle cramps.
The patient was referred to an endocrine surgeon in September 2019 for resection of her mediastinal mass, and underwent robotic-assisted right posterior mediastinal parathyroidectomy in October 2019. Pathology results demonstrated a 2.5×2.3×2.0 cm mass with histological results consistent with grade I neuroendocrine tumor (Figures 1–3). The Ki-67 index was 2%. Stains for Cam 5.2, CD56, chromogranin (Figure 4), and synaptophysin were positive, which confirmed neuroendocrine histology. Additional stains for PTH and TTF-1 were negative, excluding a primary parathyroid neoplasm (Figure 5). Negative stains for S100, SOX-10, and PAX-8 also excluded thyroid medullary carcinoma (Figure 5). One mediastinal lymph node was found to have malignant neuroendocrine histology as well, matching the tissue in the parathyroid mass. Thus, a metastatic neuroendocrine tumor with low-grade cytomorphology and a low mitotic index was diagnosed (Figure 6).
After surgery, the patient’s PTH level was persistently elevated despite the absence of parathyroid glands. Two days after parathyroidectomy, the PTH level was 221 pg/mL. The calcium level remained elevated as well, at 10.9 mg/dL, demonstrating a lack of negative feedback effect. The PTHrP level was evaluated owing to concern for paraneoplastic syndrome of malignancy. The PTHrP level was 6 pg/mL, which was within the reference range. A second technetium 99 sestamibi nuclear scan was performed and did not demonstrate any evidence of hyperfunctioning parathyroid glands or exogenous parathyroid tissue (Figure 7). The patient underwent dotatate positron emission tomography/computed tomography (PET/CT) to evaluate for residual neuroendocrine tumor, which demonstrated increased fluorodeoxyglucose (FDG) avidity in mediastinal, intra-abdominal, and para-aortic lymph nodes (Figures 8–11). The patient was still endorsing intermittent diarrhea and flushing even after the neuroendocrine tumor had been removed, as well as the manifestations of hypercalcemia. It was conjectured that the occult neuroendocrine tumor noted on dotatate PET/CT was producing exogenous PTH, given that all parathyroid glands had been excised. The patient was subsequently started on octreotide intramuscular injections of 30 mg every 4 weeks in January 2020 to treat her metastatic low-grade neuroendocrine tumor.
After this intervention, the patient reported that her diarrhea and flushing subsequently resolved, and her fatigue, bone pain, anxiety, palpitations, and memory deficits also improved. Laboratory evaluation demonstrated a reduction in PTH level to 86 pg/mL within 8 months after treatment of octreotide, with her calcium down-trending to 10.0 mg/dL as well. The patient’s chromogranin A levels fell from 190 µG/L, obtained the week prior to her neuroendocrine tumor resection, to 152 µG/L (normal) 1 year later. It remained at this level (153 µG/L) as of July 2021, while she continued octreotide injections. Serum serotonin levels were also evaluated prior to resection (108 ng/mL) and urine 5-HIAA levels were evaluated prior to treatment (0.9 mg/24 h), which were both within the reference range. Plasma metanephrines checked in January 2020 prior to treatment with octreotide were slightly above normal, at 60 pg/mL, but total plasma metanephrines on fractionation obtained on the same day were within normal limits (149 pg/ mL). Plasma catecholamines, dopamine, epinephrine, growth hormone, serum AM cortisol, FSH, LH, prolactin, and insulin-like growth factor-1 were all within the reference range. CEA was mildly elevated at 3.4 ng/mL. Urine calcium was within range, at 69 mg/24 h, as was calcium to creatinine ratio (84 mg/g of creatinine). Dotatate PET/CT in December 2020 demonstrated regression in activity of the neuroendocrine tumor from the dotatate PET/CT obtained 1 year prior. This finding was consistent with the improvement in the patient’s symptoms and reduction in both PTH and calcium levels after initiation of treatment with octreotide.
The patient underwent PGL next-generation sequencing to evaluate her risk for developing paragangliomas, pancreatic neoplasms, and pheochromocytoma. Given parathyroid tumors are commonly associated with underlying genetic syndromes (ie, MEN-1), we wanted to rule out other etiologies of this neuroendocrine tumor beyond recurrence alone of her carcinoid tumor from 10 years prior. No clinically significant variants were detected. The patient has remained on octreo-tide for 2 years and remains asymptomatic.
Discussion
Neuroendocrine tumors, which are low-grade, well-differentiated neuroendocrine malignancies, are a relatively rare and heterogenous tumor type. They comprise only 2% of all malignancies and have a prevalence of approximately 1 in 200 000 annually in the United States [5]. Neuroendocrine tumors have the capacity of producing exogenous hormones of nearly any type and can occur anywhere in the body, including the central nervous system, respiratory tract, gastrointestinal tract, thyroid, skin, breast, and urogenital system. Most are known to occur in the digestive tract (62–67%), particularly in the small bowel, and the lungs (22–27%) [5]. While any hormone can be produced, neuroendocrine tumors are generally nonproductive. When they are associated with hormone production, the most common paraneoplastic syndromes involve production of serotonin, insulin, gastrin, glucagon, somatostatin, vasoactive intestinal peptide, growth hormone, adrenal corticotropic hormone, melanocyte-stimulating hormone, pancreatic poly-peptide, calcitonin, substance P, and pancreastatin [5]. Most of these neuroendocrine tumors are not inherited, and they tend to develop sporadically in people with no family history of neuroendocrine tumors.
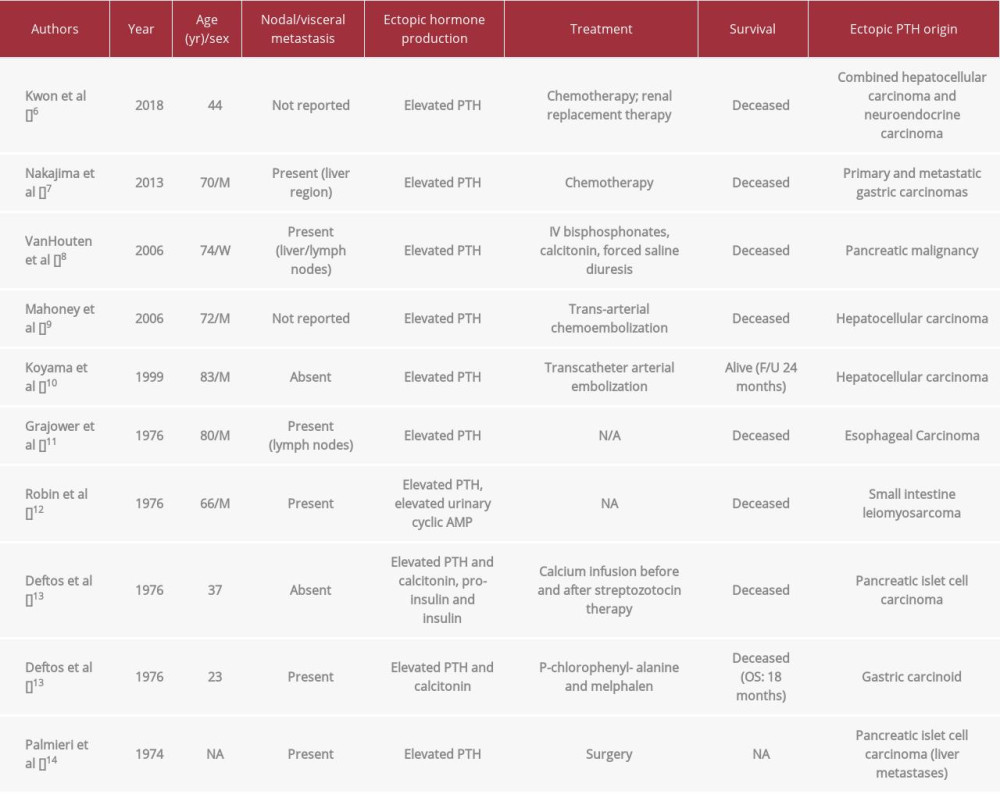
In our patient’s case, the neuroendocrine tumor was determined to be producing PTH as opposed to another typically associated neuroendocrine hormone as described above. PTH was the only elevated hormone after extensive evaluation of other hormone levels by the endocrinology department, and furthermore, it was the cause for her initial presentation to her primary care physician with symptoms of hypercalcemia as well as neuroendocrine manifestations. The etiology of the PTH production was confirmed by exclusion of PTHrP as the culprit, given that it was within the reference range, as well as by the exclusion of native PTH from parathyroid tissue due to the excision of the parathyroid glands and “parathyroid adenoma” (which was found to be a neuroendocrine tumor instead). A technetium 99 sestamibi nuclear medicine scan through the level of the mediastinum demonstrated no exogenous parathyroid tissue that could be producing native PTH. Finally, dotatate PET/CT confirmed the presence of a residual neuroendocrine tumor that was widely metastatic throughout the neck, chest, and abdomen. Subsequent treatment of the neuroendocrine tumor alone with 30 mg intramuscular octreo-tide injections every 4 weeks led to the resolution of the patient’s clinical symptoms, reduction in her serum calcium levels from 10.9 mg/dL to 10.0 mg/dL, reduction in her PTH level from 221 pg/mL to 86 pg/mL, and reduced activity of her neuroendocrine tumor on dotatate PET/CT 1 year later. Thus, by a diagnosis of exclusion, resolution of symptoms, and normalization of laboratory values during the treatment of only the neuroendocrine tumor, it is reasonable to conclude that this was a rare presentation of a PTH-producing neuroendocrine tumor. As described above, this is an extremely rare entity, described only in 10 cases in the literature with other gastrointestinal tumors, but almost never in a primary neuroendocrine tumor alone (Table 1; References [6–14] in Table 1 only).
Our patient had previously been diagnosed with a grade I carcinoid tumor after it was incidentally found in the pathology results from a small bowel resection. She was asymptomatic, and the likelihood of recurrence was low despite the invasion through the muscularis propria and into the serosa and mesentery. She was never treated with any adjuvant medication. Ten years later, the patient developed a low-grade metastatic neuroendocrine tumor, which was likely a recurrence of the original tumor, but was now manifesting as hypercalcemia due to aberrant PTH production. Owing to the coincidental first site of the neuroendocrine tumor recurrence being discovered as a mass at the level of the parathyroid glands, the site of PTH production was originally mistaken as a productive adenoma arising from the parathyroid glands. Surgical resection was pursued, demonstrating the true nature of the pathology underlying the parathyroid mass, which was histo-logically of neuroendocrine origin. The pathology further indicated the metastatic nature of this unexpected tumor, as it described invasion into a local parathyroid lymph node as well. No parathyroid tissue with PTH positivity was noted. Negative stains for PTH and TTF-1 confirmed the tumor did not arise from parathyroid origin. S100, SOX-10, and PAX-8 stains which were negative, also excluded medullary thyroid carcinoma as the origin of the neuroendocrine malignancy. PTH levels remained elevated, and the patient remained symptomatic with fatigue, memory loss, anxiety, and bone pain, despite complete parathyroid resection and a negative sestamibi nuclear scan that excluded exogenous parathyroid tissue as the culprit. With the help of a dotatate PET scan, residual neuroendocrine metastases were discovered and the suspicion of the neuroendocrine tumor being the origin of exogenous PTH production was further elevated. Treatment of the neuroendocrine tumor with octreotide subsequently led to a fall in the level of intact PTH and resolution of clinical manifestations and laboratory values of hypercalcemia and hyperparathyroidism.
From a review of the literature from 1974 through 2018, we found that neuroendocrine tumors producing native PTH have been reported in 4 cases. Of all tumors described that produced exogenous PTH, including the 4 neuroendocrine tumors mentioned above, only 10 cases have been documented (Table 1). These case reports of neuroendocrine malignancies producing PTH, also described hypercalcemia in these patients as a consequence of exogenous PTH that was difficult to control and led to significant impacts on morbidity and mortality. The authors reported that due to complications of hypercalcemia, including multi-organ failure, all patients described had a shorter overall survival compared with their counterparts who had non-PTH producing neuroendocrine malignancies. Of the 10 patients described in the literature, 8 were known to be deceased at the time of this report, despite their diagnoses being within 2 to 44 years of this description and mean age being 58 years on diagnosis. Three of the 10 were under age 45 years at diagnosis, yet they died from the disease within 18 to 24 months on average. This demonstrates the increased impact on morbidity that hypercalcemia can have on a clinical course, even in the setting of a low-grade neuroendocrine malignancy. While already rare, our case is additionally unique as none of the reports above documented a neuroendocrine tumor arising from the parathyroid gland.
As described above, the literature indicates that paraneoplastic syndromes associated with exogenous PTH carry a poor prognosis. Treatment modalities such as surgical resection and trans-catheter arterial embolization and chemotherapy to eradicate single neuroendocrine lesions have done little to increase the rate of survival in these patients, likely due to the presence of occult metastases. A disease-specific diagnostic tool such as the dotatate PET/CT has become vital for the evaluation of the disease burden in neuroendocrine tumors, especially if a paraneoplastic syndrome is present. Early identification of metastatic spread has aided in early intervention with systemic treatments and longer disease-free survival [15]. Systemic interventions also reduce the pathologic effects of malignant hormone production and reduce rates of organ failure [15].
In our case, the persistently elevated calcium and PTH levels after surgical removal of the parathyroid glands and neuroendocrine tumor were investigated with the dotatate PET/CT. It demonstrated multiple sites of occult metastasis. This led to our initiation of systemic treatment with octreotide within weeks of diagnosis. At the time of this writing, the patient remains on monthly octreotide injections with sustained normalization of calcium and PTH levels, and with resolution of both her neuroendocrine symptoms and clinical manifestations of hypercalcemia. She remains on vitamin D replacement as well as raloxifene, given she has been resistant to osteoporosis management with bisphosphonates. She is followed closely by endocrinology and oncology staff, and she has not had evidence of organ dysfunction or fracture since diagnosis.
Conclusions
Neuroendocrine cancers are able to produce intact PTH leading to paraneoplastic syndromes and hypercalcemia. A diagnosis in these patients may initially be difficult given the rarity of intact PTH production arising from malignancy and the lack of knowledge surrounding the subject. It is important to consider all the potential causes of disordered hypercalcemia upon initial evaluation of a patient and to rule out malignant PTH production through the evaluation of PTH levels, PTHrP, serum phosphorus, serum vitamin D, and technetium 99 sestamibi nuclear scans for exogenous parathyroid tissue. As evident in our case, neuroendocrine tumors should be considered in the differential diagnosis in patients with hypercalcemia of unclear origin. Evaluation with a dotatate PET scan can be very helpful in diagnosing metastatic disease. Early identification and management of these patients is essential to address the underlying cause of paraneoplastic syndromes, reduce morbidity from exogenous hormone production, and optimize overall survival.
Figures
References:
1.. Kaltas GA, Besser GM, Grossman AB, The diagnosis and medical management of advanced neuroendocrine tumors: Endocr Rev, 2004; 25(3); 458-511
2.. Mundy GR, Edwards JR, PTH-related peptide (PTHrP) in hypercalcemia: J Am Soc Nephrol, 2008; 19(4); 672-75
3.. Kandil E, Noureldine S, Khalek MA, Ectopic secretion of parathyroid hormone in neuroendocrine tumor: A case report and review of the literature: Int J Clin Exp Med, 2011; 4(3); 234-40
4.. Newman SK, PTH versus PTHrP – small differences, big implications: Clin Corr, 2016 Available from: http://www.clinicalcorrelations.org/2016/03/03/pth-versus-pthrp-small-differences-big-implications/
5.. Oronsky B, Ma P, Morgensztern D, Carter C, Nothing But NET: A review of neuroendocrine tumors and carcinomas: Neoplasia, 2017; 19(12); 991-1002
6.. Kwon HJ, Kim JW, Kim HY, Combined hepatocellular carcinoma and neuroendocrine carcinoma with ectopic secretion of parathyroid hormone: A case report and review of the literature: J Pathol Transl Med, 2018; 52(4); 232-37
7.. Nakajima K, Tamai M, Okaniwa S, Humoral hypercalcemia associated with gastric carcinoma secreting parathyroid hormone: A case report and review of the literature: Endocr J, 2013; 60(5); 557-62
8.. VanHouten JN, Yu N, Rimm D, Hypercalcemia of malignancy due to ectopic transactivation of the parathyroid hormone gene: J Clin Endocrinol Metab, 2006; 91(2); 580-83
9.. Mahoney EJ, Monchik JM, Donatini G, De Lellis R, Life-threatening hyper-calcemia from a hepatocellular carcinoma secreting intact parathyroid hormone: Localization by sestamibi single-photon emission computed tomo-graphic imaging: Endocr Pract, 2006; 12(3); 302-6
10.. Koyama Y, Ishijima H, Ishibashi A, Intact PTH-producing hepatocellular carcinoma treated by transcatheter arterial embolization: Abdom Imaging, 1999; 24(2); 144-46
11.. Grajower M, Barzel US, Ectopic hyperparathyroidism (pseudohyperparathyroidsim) in esophageal malignancy. Report of a case and a review of the literature: Am J Med, 1976; 61(1); 134-35
12.. Robin NI, Siegel L, Hawker C, Magida MG, Hypercalcemia and metastatic intestinal leiomyosarcoma: A case of ectopic parathyroid hormone production: Connecticut Med, 1976; 40(9); 609-11
13.. Deftos LJ, McMillan PJ, Sartiano GP, Simultaneous ectopic production of parathyroid hormone and calcitonin: Metab, 1976; 25(5); 543-50
14.. Palmieri GM, Nordquist RE, Omenn GS, Immunochemical localization of parathyroid hormone in cancer tissue from patients with ectopic hyper-parathyroidism: J Clin Invest, 1974; 53(6); 1726-35
15.. Ter-Minassian M, Zhang S, Brooks N, Association of tumor progression end points and overall survival in patients with advanced neuroendocrine tumors: Oncologist, 2017; 22(2); 165-72
Figures
In Press
06 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.942937
12 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943244
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943275
13 Mar 2024 : Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.943411
Most Viewed Current Articles
07 Mar 2024 : Case report
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133
10 Jan 2022 : Case report 
DOI :10.12659/AJCR.935263
Am J Case Rep 2022; 23:e935263
19 Jul 2022 : Case report
DOI :10.12659/AJCR.936128
Am J Case Rep 2022; 23:e936128
23 Feb 2022 : Case report
DOI :10.12659/AJCR.935250
Am J Case Rep 2022; 23:e935250