19 August 2020: Articles 
Hemophagocytic Lymphohistiocytosis and Relapsing Polychondritis with Acute Myelogenous Leukemia: Case Report and Review of the Literature
Challenging differential diagnosis, Rare disease, Rare coexistence of disease or pathology
Hamza M. Alsaid1ABCDEF*, Adnan A.M. Wahdan2BCDEFG, Ihab N. Tahboub3ABCDEF, Nour M. Almakadma2ABCDEFDOI: 10.12659/AJCR.925287
Am J Case Rep 2020; 21:e925287






Abstract
BACKGROUND: This case report describes rare disease entities with possible associations that include relapsing polychondritis, a rare disease with systemic manifestations characterized by bouts of inflammation in hyaline cartilage in multiple body sites, and hemophagocytic lymphohistiocytosis (HLH), another potentially life-threatening condition that occurs due to erratic activation of the immune system accompanied by pancytopenia. Both diseases constitute a real challenge to diagnose and treat. These entities, their associations, and treatment protocols and prognosis for them are highlighted.
CASE REPORT: A 16-year-old female presented with features and complications of both relapsing polychondritis (RP) and HLH including costochondritis, fever, splenomegaly, thrombocytopenia, and anemia. After admission to the intensive care unit, symptomatic management included paracetamol, intravenous fluids, prednisolone 60 mg orally, intravenous immune globlulin, and warfarin. Unfortunately, the patient developed acute myelogenous leukemia (FAB AML M5b) after a period of remission and died due to sepsis and multiorgan failure.
CONCLUSIONS: HLH and RP are two rare diseases that can present together. Whether this malignant process (AML) is a cause or a result of these diseases is unknown. In the case presented here, the patient developed features of AML after a period of remission from RP and HLH. This case report may provide perspective on diagnosis and treatment for clinicians faced with similar patients.
Keywords: Leukemia, Myeloid, Acute, Lymphohistiocytosis, Hemophagocytic, Polychondritis, Relapsing, Adolescent, Multiple Organ Failure, Pancytopenia
Background
Relapsing polychondritis (RP) is a rare autoimmune disease that has systemic manifestations. It is characterized by recurrent bouts of inflammation that involves cartilage throughout the body, but mainly in the nose, ear, respiratory tract, and cardiovascular system [1]. Incidence of RP is estimated to be 3.5 cases per million/year, with a nearly equal frequency in males and females [1]. In a population-based cohort study in the UK, the mean ages at diagnosis were 55 and 51 years in males and females, respectively; median time from symptoms appearance to diagnosis was about 1.9 years [2].
The exact pathogenesis of RP is not yet fully understood, however, it is believed that it represents an autoimmune response resulting in cartilage matrix destruction by proteolytic enzymes due to tissue infiltration by macrophages, neutrophils, CD4-positive T lymphocytes, and autoantibodies directed against type II collagen in hyaline cartilages [3].
RP can have various and unpredictable clinical presentations. Arthritis seems to be the most consistent and common manifestation. However, more than half of patients show no evidence of chondritis, and a concomitant autoimmune disease may be present in one-third of patients [4].
Various triggers may lead to RP, such as trauma (e.g. ear piercing), infections such as
Hemophagocytic lymphohistiocytosis (HLH) is another rare, life-threatening clinical syndrome caused by an inflammatory signal error, usually originating from abnormalities in natural killer (NK) and/or cytotoxic T cells, which lead to uncontrolled and ineffective immune system response, eventually culminating in serious elevations in many inflammatory cytokines if left untreated [6,7]. HLH is associated with several conditions, such as autoimmune diseases, infections, certain neoplasms, medications, and well-recognized hereditary diseases [8]. The syndrome also has many clinical features, which include progressive organ enlargement, worsening cytopenias, ferritin level elevations out of proportion to the degree of the inflammation, worsening liver function tests, and clotting disorders [8]. In HLH, defective killing of pathogens and cancer cells not only renders immune cells unable to kill their target, but also leads to a dangerous state of systemic hypercytokinemia, which is well-demonstrated in genetic forms of this disease [6].
HLH can be classified into two major groups: familial (primary), and acquired (secondary) [6]. Secondary HLH initially was described as related to viral infections following organ transplantations; virus-associated HLH also has been reported [9]. Other important secondary causes of HLH include malignancies such as leukemia. O’Brien and colleagues described three cases of HLH secondary to B-ALL [10].
The case reported here illustrates a possible association between RP and HLH in a 16-year-old female patient who presented with features of both disease entities and had developed acute myelogenous leukemia (AML) during the course of her illness. This unusual presentation and the approach used for treatment are described.
Case Report
A 16-year-old female presented to the Internal Medicine and Rheumatology Department at Palestine Medical Complex with a complaint of a 2-week history of fever (39°C, 102.2°F).
The patient was in her usual state of health until 14 days prior her admission, when she started to experience fever. She also complained of generalized weakness, joint pain, mainly in her elbows and back, but no swelling or stiffness. The patient denied having skin rash, abdominal pain, headaches, visual disturbances, changes in urinary or bowel habits, or change in weight. She also denied ill contacts.
On physical examination, the patient looked ill and fatigued. Her ears, except the earlobes, were red and swollen, suggesting auricular perichondritis (Figure 1A, 1B). Musculoskeletal examination revealed joint line tenderness involving both elbows, the back, and multiple costochondral joints. There were no limitations of active or passive range of motion nor any joint swelling or redness. Cardiovascular and respiratory exam were within normal limits with normal chest shape and contour and no murmurs, added sounds, wheeze or crackles. Other than splenomegaly, there was no tenderness, rigidity, or palpable masses on abdominal examination.
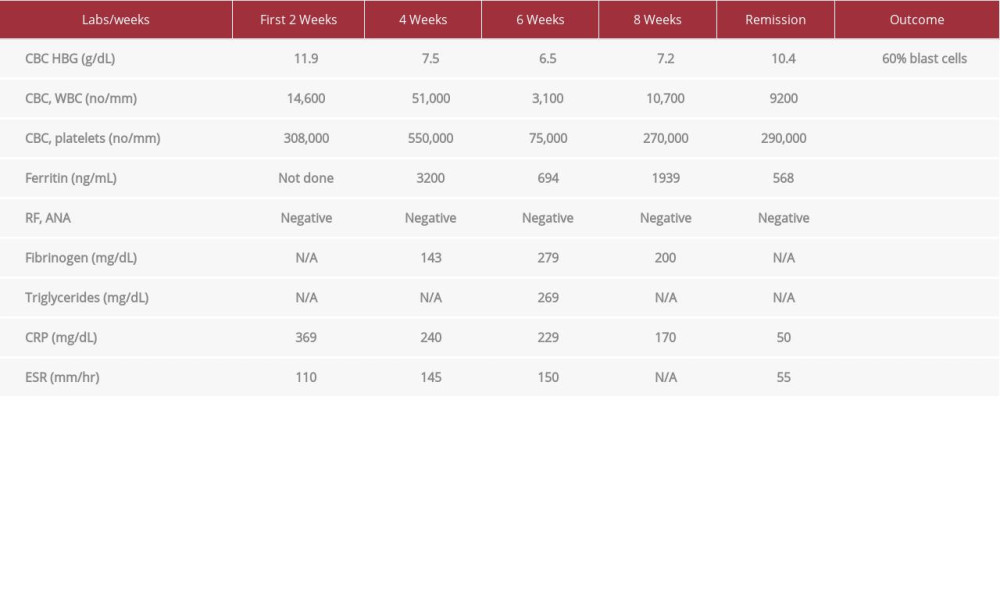
The patient’s vital signs on admission were as follows: temperature, 39°C; pulse rate, 102 beats per minute (BPM); blood pressure, 102/80 mmHg; respiratory rate, 18 respirations per minute; and oxygen saturation, 98%. Results of blood testing were as follows: hemoglobin (HGB), 11.9 g/dL; mean corpuscular volume, 79.2 mm3; platelets, 308,000/mm3; white blood cell count, 14,600/mm3; C-reactive protein (CRP), 369 mg/L; erythrocyte sedimentation rate (ESR), 110 mm/hr; and lactate dehydrogenase, 561 U/L (Table 1).
The patient was treated as a suspected case of sepsis upon admission, for which she received broad spectrum antibiotics, in addition to supportive treatment consisting of intravenous fluids and paracetamol.
Further evaluation during the patient’s hospitalization included testing for
During hospitalization, the patient underwent various imaging studies. Abdominal ultrasound revealed an enlarged spleen of 15 cm (in the greatest dimension) and a normal-size liver with no abnormalities. Whole-body computed tomography scan showed an enlarged spleen and mesenteric lymph node enlargement (Figure 2A, 2B). Echocardiography showed significant apical hypokinesia, with moderate pulmonary hypertension of 50 mmHg. Echocardiography also showed moderate tricuspid regurgitation with a mobile echo-dense structure on the pulmonic valve, mostly correlating with a vegetation (Figure 3).
Blood film showed microcytic hypochromic red blood cells with Rouleax formation, leukocytosis with segmented neutrophils and occasional monoblasts, for which the patient was transferred to a hematology/oncology facility to rule out acute myelogenous leukemia (AML). During her stay in the oncology center, she continued to have fever along with tachycardia of 110 bpm and hypotension of 90/58 mmHg. Lab tests were repeated and showed leukocytosis of 51,000/mm3 with absolute neutrophil count of 36,000/mm3. HBG dropped to 7.5 g/dL. CRP and ESR were 240 mg/L and 145 mm/hr, respectively. The patient was started on broad spectrum antibiotics again. Results of subsequent urine and blood cultures were negative. The patient deteriorated and was transferred to the intensive care unit (ICU) for further evaluation and management.
During the patient’s stay in the ICU, her laboratory test results were as follows: ferritin, 3200 ng/mL, fibrinogen, 1.43 mg/dL, total serum bilirubin, 2.7 mg/dL; direct serum bilirubin, 1.2 mg/dL; and LDH, 583 U/L. Testing for cytoplasmic anti-neutrophil cytoplasmic antibodies (C-ANCA), perinuclear ANCA, antinuclear antibodies,RF, Anti-Jo, anti-Sjögren’s-syndrome-related antigen A and B antibodies, anti-double-stranded DNA, anti-smith, and anti-scl-70 antibodies was negative (Table 1).
In the ICU, the patient developed palpable purpuric and bullous skin lesions with irregular borders on the extensor aspect of the right elbow. (Figure 4). Rheumatology team opinion was sought, a skin biopsy of the lesion was done, and showed epidermal edema with diffuse interstitial neutrophilic infiltration in the papillary dermis and the reticular dermis, without evidence of vasculitis.
A final diagnosis of RP with HLH was made, and the patient was treated as such. Acute hospital ICU management consisted of treatment of symptoms with paracetamol, IV fluids, and 2 units of packed RBCs. The patient also received prednisolone 60 mg orally, IVIG, and hydroxyurea. The latter drug was added because the patient developed marked leukocytosis and it was discontinued immediately after resolution. Based on evaluation by the cardiology team, the patient also received warfarin 5 mg per day and furosemide 40 mg per day due to heart failure and apical ballooning. After her condition had stabilized, the patient was maintained on cyclosporine 100 mg twice daily and prednisone 60 mg. She went into remission but unfortunately relapsed 4 weeks later. Another bone marrow examination was performed and a diagnosis of AML was established, based on the predominance of mono-blasts and promonocytes, which accounted for 60% of all cells (Table 1). The test also showed decreased granulocytes with dysplastic and hypogranular cytoplasm; Pelger-Huet cells; and decreased erythrocytes and megakaryocytes with M: E ratio of 5: 1. Based on results of flow cytometry, the patient’s disease was designated FAB AML M5b, AML with myelodysplasia-related changes. It is noteworthy that, before this diagnosis, serial bone marrow biopsies failed to show evidence of significant blastic changes (>20% blasts). The patient was treated with cytarabine 100 mg/m2 (160 mg) every 12 hours, over 30 minutes and received a total of 12 doses over 6 days). She also received allopurinol. Because of heart failure, the patient was ineligible for doxuribicin, and therefore, the treatment protocol for AML could not be completed, she died 3 weeks later due to
Discussion
This case report illustrates a constellation of rare diseases in a 16-year-old female patient. According to reports from case studies and one case series, the mean age of onset of RP in children is 15.1 years [11], and ranges from 1.7 months to 17 years [12]. The patient described here presented with common manifestations of RP: arthritis and auricular perichondritis [4]. In the pediatric population, the symptoms and tissue involvement are comparable to those reported in adults, but pediatric patients tend to have more severe manifestations. However, the reported cases in the pediatric population constitute less than 5% of those reported in all age groups [11,12].
The etiology of RP is not well-known [13]; however, many factors can trigger RP, including traumatic piercing of cartilage (e.g. ears, nose), infections (e.g.
RP is often difficult to diagnose because of the lack of confirmative diagnostic tests and its relapsing nature and heterogeneous symptoms. In fact, diagnosis mainly relies on the clinical picture, imaging studies, and biopsy of the affected tissues [5].
Interestingly, the case reported here is the second to show a possible association between RP and HLH [14]. HLH is a disorder of immune dysfunction resulting in uncontrolled activation of macrophages and NK cells [14]. Like RP, the clinical manifestations and laboratory test results for HLH are nonspecific. In fact, HLH may also share clinical and laboratory manifestations of its common triggers, such as sepsis and lymphoma [15]. Therefore, Hunter et ale developed criteria for diagnosis of HLH. At least five of the following should be present: fever; cytopenias involving at least two lines; increased triglycerides and/or decreased fibrinogen; splenomegaly; ferritin elevation to levels more than 500 ng/mL; absence or decreased activity of NK cells; bone marrow, liver or lymph node samples showing hemophagocytosis; and elevations of soluble interleukin (IL)-2 receptor-α levels >2400 U/mL (Table 2) [16]. The patient in this case presented with fever, splenomegaly, leukocytopenia and erythrocytopenia, hypertriglyceridemia and high levels of ferritin, and thus fulfilled Hunter’s criteria for HLH.
Despite its name, microscopic visualization of hemophagocytosis is simply one of the possible diagnostic criteria for HLH. It does not necessarily need to be present to establish the diagnosis. In fact, hemophagocytosis is often cyclical, thus a given biopsy specimen may yield negative results at first examination [17].
In this case, unfortunately, sIL-2R levels and NK activity were not measured because of issues with the patient’s insurance. Moreover, these tests are not available in everyday practice [15].
All of the findings of HLH are related to hypercytokinemia and infiltration of organs with activated lymphocytes and histiocytes [7], Fever is related to high IL-1 and IL-6 levels, whereas pancytopenia is related to high levels of TNF-α and interferon-gamma, in addition to hemophagocytosis itself [18].
During the course of her illness, the patient here unfortunately developed AML. This actually makes it the first reported case with a combination of RP, HLH, and AML. In the literature, it has been reported that HLH can be secondary to a malignancy. O’Brien and colleagues described three cases of HLH secondary to B-acute lymphoblastic leukeia (ALL) [10], while another article reported a child with precursor B- ALL who was diagnosed with HLH after experiencing splenomegaly and refractive thrombocytopenia during induction chemotherapy [19]. In addition, another case report of a patient with neurofibromatosis also shed light on a possible association between Jjvenile myelomonocytic leukemia and HLH as it presented with features of the latter disease [20].
An association between RP and myelodysplastic syndromes (MDS) has been reported, as MDS has been documented in one-third of patients with RP [21]. Erden and colleagues reported a case of RP in which the patient developed MDS 14 years after the initial diagnosis of RP [22]. According to Mekinian et al., the reported time interval from development of RP to MDS was 8.4 months [23]. In the case presented here, the patient developed AML about 4 months after the initial diagnosis of RP.
Treatment of RP depends on the acuteness and severity of the disease manifestations. Patients with mild acute disease are usually treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and prednisone, whereas those with more severe manifestations, such as airway compromise, may require higher doses of prednisone or even IV pulse methyl-prednisone [1]. Many other newer treatments also have been deployed for RP, such as immunosuppressants, biologic agents, antitumor necrosis factor-alpha (TNF-α) agents, and IL-1 receptor antagonist (Anakinra) [5]. Erden and colleagues reported a remission of RP after successful treatment of MDS with the hypomethylating agent azacytidine [22].
As for RP treatment in the pediatric group in particular, in a case series reported by Belot, patients mainly received corticosteroids, NSAIDs, and immunosuppressants [12]. In another case reported by Buonuomo, Anakinra was used and showed good results in slowing progression of the disease [24].
In another case report, RP has been described as a presenting feature of AML, with resolution of features of RP after induction chemotherapy with idarubicin and cytarabine [25]
The goal of HLH treatment is to suppress life-threatening inflammation. In 1994, The Histiocyte Society organized the first treatment protocol for HLH (HLH-94), which dramatically increased the survival rate to 54%t [26]. Under the HLH-94 Protocol, treatment consists of Induction therapy using weekly etoposide and hydrocortisone followed by hematopoieitic stem cell transplantation if the patient is not improving. Improving patients are weaned off therapy. Patients with central nervous system disease are given intrathecal methotrexate and hydrocortisone [26].
A newer protocol was developed in 2004. The major modification consisted of adding cyclosporine to the induction phase of treatment [27]. That protocol was followed in the case presented here and the patient received cyclosporine and prednisone.
Regarding prognosis, with RP, it usually is good prognosis, but it can be life-threatening in cases with tracheal and cardiac involvement [5]. As for HLH, it has been stated that in pediatric patients, it can be fatal, as up to 95% of affected children with the disease who are not treated succumb [28].
Conclusions
In summary, this case report presents a rare constellation of diseases: RP, HLH, and AML. They occurred in a pediatric patient who initially developed features of both RP and HLH, followed by a period of remission after treatment with cyclosporine, IVIG, and prednisone, and eventually developed AML M5b a few weeks later. We believe that the challenging part of the case report lies in the rarity of the diseases it encompasses, and the difficulty in assessing associations between them. We hope this case report will be of benefit to clinicians, in terms of guiding diagnosis and treatment in future similar patients.
Figures
References:
1.. Longo L, Greco A, Rea A, Relapsing polychondritis; a clinical update: Autoimmunity Re, 2016; 15(6); 539-43
2.. Hazra N, Dregan A, Charlton J, Incidence and mortality of relapsing polychondritis in the UK: A population-based cohort study: Rheumatology, 2015; 54(12); 2181-87
3.. Vitale A, Sota J, Rigante D, Relapsing polychondritis: An update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives: Curr Rheumatol Rep, 2016; 18(1); 3
4.. Puéchal X, Terrier B, Mouthon L, Relapsing polychondritis: Joint Bone Spine, 2014; 81(2); 118-24
5.. Mathian A, Miyara M, Cohen-Aubart F, Relapsing polychondritis: A 2016 update on clinical features, diagnostic tools, treatment and biological drug use: Best Practice Res Clin Rheumat, 2016; 30(2); 316-33
6.. Ünal S, Hemophagocytic lymphohistiocytosis: an update to diagnosis and management: Acta Medica, 2014; 45(1); 29-34
7.. Vaiselbuh SR, Bryceson YT, Allen CE, Updates on histiocytic disorders: Pediatr Blood Cancer, 2014; 61(7); 1329-35
8.. Janka GE, Familial and acquired hemophagocytic lymphohistiocytosis: Eur J Pediat Feb 1, 2007; 166(2); 95-109
9.. Risdall RJ, McKenna RW, Nesbit ME, Virus-associated hemophagocytic syndrome A benign histiocytic proliferation distinct from malignant histiocytosis: Cancer, 1979; 44(3); 993-1002
10.. O’Brien MM, Lee-Kim Y, George TI, Precursor B-cell acute lymphoblastic leukemia presenting with hemophagocytic lymphohistiocytosis: Pediatr Blood Cancer, 2008; 50(2); 381-83
11.. Lin DF, Yang WQ, Zhang PP, SAT0262 7 cases of Chinese juvenile relapsing polychondritis and systemic review: Ann Rheumat Dis, 2016; 75(2); 763
12.. Belot A, Job-Deslandre C, Boudjemaa S, Relapsing polychondritis: A pediatric series of ten patients: Pediatr Rheumatol, 2008; 6(Suppl.1); P235
13.. Knipp S, Bier H, Horneff G, Relapsing polychondritis in childhood – case report and short review: Rheumatol Int, 2000; 19(6); 231-34
14.. Monaco WE, Field CJ, Taylor TH, A case of relapsing polychondritis mimicking hemophagocytic lymphohistiocytosis after Propionibacterium acnes infection: J Clin Rheumat, 2019; 25(5); e75-77
15.. Larroche C, Hemophagocytic lymphohistiocytosis in adults; diagnosis and treatment: Joint Bone Spine, 2012; 79(4); 356-61
16.. Henter JI, Tondini C, Pritchard J, Histiocyte disorders: Critical Rev Oncol Hematol, 2004; 50(2); 157-74
17.. Hsi ED: Hematopathology: A volume in foundations in diagnostic pathology series, 2007, London, UK, Churchill Livingstone
18.. Janka GE, Lehmberg K, Hemophagocytic syndromes – an update: Blood Rev, 2014; 28(4); 135-42
19.. Kelly C, Salvi S, McClain K, Hayani A, Hemophagocytic lymphohistiocytosis associated with precursor B acute lymphoblastic leukemia: Pediatr Blood Cancer, 2011; 56(4); 658-60
20.. Shin HT, Harris MB, Orlow SJ, Juvenile myelomonocytic leukemia presenting with features of hemophagocytic lymphohistiocytosis in association with neurofibromatosis and juvenile xanthogranulomas: J Pediatr Hematol Oncol, 2004; 26(9); 591-95
21.. Okamoto T, Okada M, Mori A, Correlation between immunological abnormalities and prognosis in myelodysplastic syndrome patients: Int J Hematol, 1997; 66(3); 345-51
22.. Erden A, Bilgin E, Kılıç L, Remission of relapsing polychondritis after successful treatment of myelodysplastic syndrome with azacitidine: A case and review of the literature: Drug Metab Personalized Ther, 2018; 33(2); 105-8
23.. Mekinian A, Grignano E, Braun T, Systemic inflammatory and auto-immune manifestations associated with myelodysplastic syndromes and chronic myelomonocytic leukaemia: A French multicentre retrospective study: Rheumatol, 2016; 55(2); 291-300
24.. Buonuomo PS, Bracaglia C, Campana A, Relapsing polychondritis; New therapeutic strategies with biological agents: Rheumatol Int, 2010; 30(5); 691-93
25.. Hsu YT, Chen TY, Relapsing polychondritis as a presenting feature of acute myeloid leukaemia: Br Journal Haematol, 2018; 180(4); 472
26.. Henter JI, Samuelsson-Horne A, Arico M, Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation: Blood, 2002; 100(7); 2367-73
27.. Henter JI, Horne A, Aricó M, HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis: Pediatr Blood Cancer, 2007; 48(2); 124-31
28.. , Histiocyte Society. Treatment protocol of the second international HLH study 2004 (HLH– 2004). Adapted from Henter JI, Elinder G, Ost A and the FHL Study Group of the Histiocyte Society. Diagnostic Guidelines for the Hemophagocytic Lymphohistiocytosis: Semin Oncol, 1991; 18; 29-33
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953068
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133