15 October 2020: Articles 
Torsades de Pointes During Myringotomy in a Child with Congenital Long QT Syndrome: A Case Report
Management of emergency care, Congenital defects / diseases
Melissa Coleman1ADEFG*, Jason R. Imundo2DEF, Daniel Cortez3DEF, Mark H. Cohen2E, Padmani Dhar1EF, Priti G. Dalal1ADEDOI: 10.12659/AJCR.925602
Am J Case Rep 2020; 21:e925602






Abstract
BACKGROUND: Long QT syndrome (LQTS) is an arrhythmogenic heart condition that can be congenital or acquired. Prolonged ventricular repolarizations in individuals with the disorder can cause fatal arrhythmias. Abnormal functioning of cardiac ion channels leads to arrhythmias such as torsades de pointes (TdP) and may be triggered by stress or medications. Many medications used in the perioperative period are triggers for the arrythmia.
CASE REPORT: A 7-year-old patient with known congenital LQTS type 2 presented for bilateral myringotomy and tube placement. The patient was otherwise healthy and taking propranolol daily. Preoperative midazolam was administered for anxiolysis, and induction of anesthesia was uneventful. He sustained an episode of TdP immediately following general anesthetic induction after failure of an in situ automatic implantable cardioverter-defibrillator (AICD). External defibrillation succeeded, and the patient was stabilized in the Postanesthesia Recovery Unit before transfer to the Pediatric Intensive Care Unit. Interrogation of the AICD revealed several undelivered defibrillation attempts. A chest X-ray showed an area suggestive of an epicardial electrode fracture. The following day, the AICD was replaced with no arrythmias noted. The patient had an uneventful recovery.
CONCLUSIONS: In patients with a known history of LQTS, preparation and prevention are cornerstones of anesthesia care. Minimizing the use of triggering medications and emotional stress in the perioperative period, combined with ready equipment and medications to respond to arrythmias, are essential. In children, there is a greater chance of lead fracture and resulting device failure. Preoperative history of device function or interrogation of the AICD and possibly a chest X-ray are essential to ensure the integrity of the leads.
Keywords: Anesthesia, General, Anesthesia, Inhalation, Long QT Syndrome, Pediatrics, Torsades de Pointes, Child, Electrocardiography
Background
Congenital long QT syndrome (LQTS) results from mutations in cardiac ion channels [1]. On electrocardiogram (ECG), prolonged QT intervals and T-wave abnormalities from extended ventricular repolarization are hallmarks of the disease. The condition can present as recurrent episodes of syncope, seizure-like episodes, or cardiac arrest. If untreated, sudden death may result from torsades de pointes (TdP) degenerating to ventricular fibrillation [2]. We present a case of a 7-year-old child with known congenital LQTS and an automatic implantable cardioverter-defibrillator (AICD)
Case Report
A 7-year-old child (weight 26 kg, height 124 cm) presented for myringotomy and ear tube placement. He had a past history of LQTS type 2, and an epicardial dual-chamber/epicardial patch AICD had been implanted 2 years prior without any anesthesia complications. No history of the AICD firing was noted during his cardiology follow-up since its placement. He had no other medical issues. Family history was positive for LQTS in the mother and sister. The child’s routine medications included propranolol. His preoperative vital signs were stable, and examination findings were normal. The pacemaker clinic recommended no changes to the AICD programming, and no interrogation was performed the day of surgery because use of surgical cautery was not planned. No ECG was performed preoperatively on the day of surgery. His ECG from more than 1 year ago revealed normal sinus rhythm with QT and QTc intervals of 482 and 559 milliseconds, respectively. All previous ECGs had a QTc interval of greater than 500 milliseconds. An echocardiogram, approximately 1 year ago, revealed normal biventricular size and function.
The child received oral midazolam for preoperative anxiolysis. In the operating room, following application of standard American Society of Anesthesiologists monitoring, inhalational induction was achieved with a 50: 50 mixture of nitrous oxide/oxygen and sevoflurane. The patient remained in normal sinus rhythm (Figure 1A). Shortly after anesthesia induction, the ECG monitor revealed a pattern of bigeminy (Figure 1B) and trigeminy, followed by 5 beats of ventricular tachycardia, which resolved spontaneously to normal sinus rhythm. Sevoflurane was immediately discontinued; 100% oxygen was administered in preparation of awakening the patient. External defibrillator pads were applied. Two minutes later, the ECG monitor revealed ventricular bigeminy followed by a pattern suggestive of TdP. The episode of TdP resolved spontaneously. Intravenous access was established during this episode. Laboratory results revealed a serum potassium level of 4.1 mmEq/L. Two minutes later, a sustained episode of TdP was noted (Figure 1C), and external cardiac defibrillation was successfully attempted (50 J) due to the failure of the
The cardiology team performed AICD device interrogation in the PACU. Evaluation of the device revealed 6 failed attempted therapies during this incident. The chest X-ray revealed an area suggestive of epicardial electrode fracture (Figure 2).
The child was admitted to the Pediatric Intensive Care Unit. He was monitored on telemetry, and defibrillation pads remained in place overnight. A transvenous single-chamber single-coil AICD was placed under general anesthesia the following day. The patient made an uneventful recovery and was discharged to home.
Discussion
We have presented the case of a child with congenital LQTS type 2 and an AICD device, who received anesthesia. Device malfunction resulted in cardiac arrest requiring treatment with defibrillation and intravenous magnesium therapy. This case presents 2 important learning points: (1) preoperative preparation of a child with an AICD, and (2) anesthetic management of a child with congenital LQTS.
LQTS can be congenital or acquired, although up to 30% of carriers of LQTS are asymptomatic and have a normal QT interval and Schwartz score [3]. Acquired LQTS denotes patients with normal QTc at baseline who may develop fatal arrhythmias when exposed to triggering agents. Due to the complex nature of LQTS genetics, patients may appear to have a normal phenotype unless their reserve is sufficiently depleted by stress or medications [4,5]. Medications that may induce TdP include certain antiarrhythmics, antibiotics, antiemetics, and induction agents [6]. The mutations in congenital LQTS mainly relate to dysfunctional proteins that disrupt the ion channel function or change the voltage threshold [4]. There are 15 major types of LQTS identified, but LQTS types 1, 2, and 3 are the most common [3]. Our patient was a known carrier of LQTS type 2, but any patient with latent disease may suddenly present with symptoms of LQTS due to the stress and pharmacology of an anesthetic.
Anesthesia management must include strategies for both prevention and treatment of arrhythmias that can lead to cardiac arrest. Preoperative prevention includes maintaining therapy with beta-blockers up to the day of surgery. Beta-blockers are the mainstay of prophylactic therapy for LQTS, but efficacy varies depending on the genotype. LQTS type 1 responds best to beta-blockers, which are less protective in LQTS types 2 and 3. Maintaining serum potassium levels in the high-normal range of 4.5 to 5 mEq/L may be protective, especially in patients with LQTS type 2 whose ion channel abnormality is sensitive to serum potassium levels [3,7]. Adequate premedication to reduce anxiety, and thereby sympathetic activation, is of paramount importance. Premedication with midazolam is preferred over dexmedetomidine because there are mixed reports in the literature about the effects of dexmedetomi-dine on QTc [8].
The Heart Rhythm Society, along with the American Society of Anesthesiologists, recommends communication with the heart rhythm team prior to any procedure and a thorough review of the records [9]. In patients with a well-established congenital LQTS diagnosis, a prolonged QTc is a known finding. In the above case, a preoperative ECG was not performed on the day of surgery because the patient had previously diagnosed congenital LQTS type 2 with a QTc greater than 500 milliseconds on all previous ECGs.
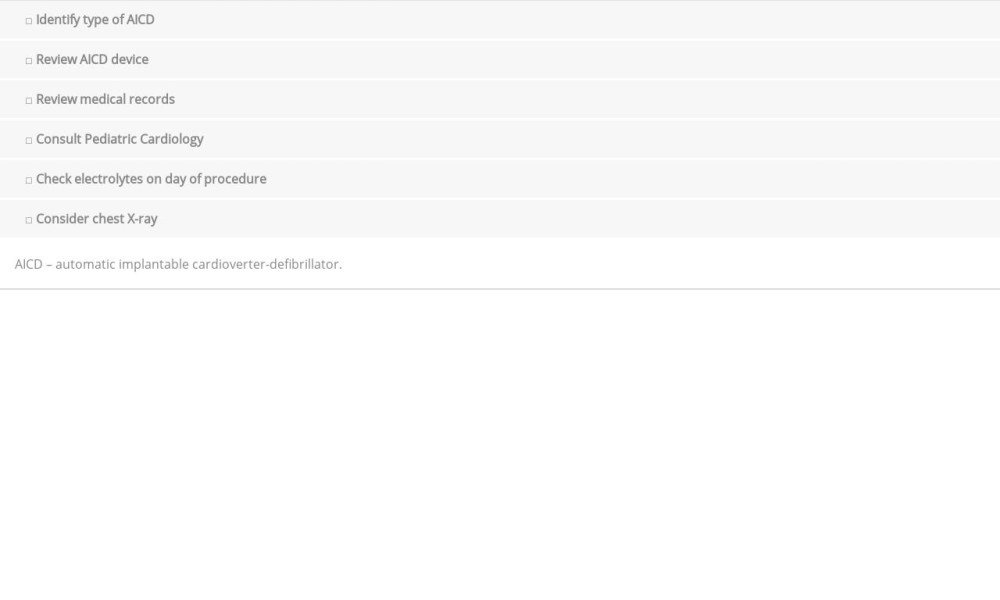
Interrogation of the device in question may or may not be necessary depending on the record of recent device checks [9]. Generally, implanted pacemakers and defibrillators are checked by technicians when electrocautery is planned because AICD tachycardia therapies would need to be turned off in this setting. Devices of pacemaker-dependent patients should be placed in an asynchronous pacing mode, preventing inhibition of their device and subsequent asystole from cautery [9]. Patients typically receive an identification card with the type of device they have (company), thus making interrogation of the device more straightforward. Ideally, a child with an AICD should have the device function confirmed before undergoing a surgical procedure. In the above case, a preoperative interrogation of the AICD would have indicated lead malfunction. There may be an increased rate of lead fracture in children compared with adults because of growth and a greater activity level [10,11]. Several strategies are used in this instance, including review of preoperative chest X-ray, recent device interrogation, and medical records. If none are available, pre-operative consultation along with a device check is warranted [9] (Table 1).
Intraoperatively, the main objectives are to prevent sympathetic excitation and to avoid factors that prolong QT interval [4]. An extensive list exists of medications that prolong the QT interval, and various drugs used in the perianesthesia period may need to be avoided [6]. In general, anesthetic agents have depressive effects on cardiovascular physiology. All volatile agents can prolong the QTc interval, although they do so to varying degrees, likely depending on the genotype of the individual [4]. Generally, patients with LQTS are not triggered by volatile agents if they are appropriately beta-blocked. Isoflurane is the agent of choice because there it may shorten QT interval [7,12]. Narcotics assist in reducing adrenergic stimulation and fentanyl is preferred, especially over sufentanil, which may increase the QTc interval [12]. Muscle relaxants such as succinylcholine and pancuronium can stimulate sympathetic responses and increase the QT interval, and therefore, they should be avoided [4]. Rocuronium and vecuronium are preferred neuromuscular blocking agents. Propofol may be beneficial because it either causes no change or shortens the QTc interval [12]. For nausea and vomiting prophylaxis, droperidol is known to prolong the QTc interval. Ondansetron is considered to be a possible risk; however, its safety profile is better [12].
If an arrhythmia is triggered, medications and equipment must be immediately available. First, discontinue the triggering agent, which during an anesthetic, is likely the volatile agent. TdP is treated with magnesium 30 mg/kg bolus over 2 to 3 minutes followed by an infusion [4,13]. In stable patients with TdP resistant to magnesium, transcutaneous or transvenous pacing can be used to increase the heart rate and shorten the QT interval [7,14]. Some LQTS patients do benefit from overdrive pacing when the TdP is induced by bradycardia or pause episodes. It is unclear, specifically, in patients with LQTS type 2 if ventricular overdrive pacing would shorten the QTc interval prophylactically. Any unstable patient should receive unsynchronized defibrillation and initiation of chest compressions. Some reports discuss using isoproterenol or dobutamine in patients with acquired LQTS who do not respond to magnesium; however, this is not indicated in patients with congenital LQTS because the increased adrenergic state may worsen the arrhythmia [12]. Any patient experiencing serious complications of LQTS should have immediate consultation and assessment with a pediatric electrophysiologist. Long-term therapy often includes AICD placement or left cardiac sympathetic denervation.
Conclusions
Although LQTS syndrome is not a common disorder, it affects about 1 in every 2000 individuals, making it a disorder the anesthesiologist may frequently encounter [3]. Beta-blockade, anxiolytics, and pain control will minimize the risk of triggering sustained perioperative arrhythmias in LQTS patients. In children, specifically, there is greater chance of lead fracture and resulting device failure. Documented history of device function or interrogation of the AICD and possibly a chest X-ray are also essential to ensure the integrity of the leads. Having magnesium and a defibrillator on standby will minimize treatment delays if a ventricular arrhythmia occurs. In patients with a known history of LQTS, preparation and prevention are the cornerstones of anesthesia care.
Figures
References:
1.. Ackerman M, The long QT syndrome: Ion channel diseases of the heart: Mayo Clin Proc, 1998; 73(3); 250-69
2.. Ackerman M, Porter C, Identification of a family with inherited long QT syndrome following a pediatric near-drowning: Pediatrics, 1998; 101; 306-8
3.. Mizsawa Y, Horie M, Wilde A, Genetic and clinical advances in congenital long QT syndrome: Circ J, 2014; 78(12); 2827-33
4.. Booker P, Whyte S, Ladusans E, Long QT syndrome and anaesthesia: Br J Anaesth, 2003; 90; 349-66
5.. Yang P, Kanki H, Drolet B, Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes: Circulation, 2002; 105(16); 1943-48
6.. Kannankeril P, Roden D, Darbar D, Drug-induced long QT syndrome: Pharmacol Rev, 2010; 62(4); 760-81
7.. Kaye AD, Volpi-Abadie J, Bensler J, QT interval abnormalities: Risk factors and perioperative management in long QT syndromes and torsades de pointes: J Anesth, 2013; 27(4); 575-78
8.. Görges M, Whyte S, Sanatani S, Changes in QTc associated with a rapid bolus dose of dexmedetomidine in patients receiving TIVA: A retrospective study: Paediatr Anaesth, 2015; 25(12); 1287-93
9.. Crossley G, Poole J, Rozner M, The Heart Rhythm Society (HRS)/ American Society of Anesthesiologists (ASA) Expert Consensus Statement on the perioperative management of patients with implantable defibrillators, pacemakers and arrhythmia monitors: Facilities and patient management: Heart Rhythm, 2011; 8(7); 1114-52
10.. Link M, Hill S, Cliff D, Comparison of frequency of complications of implantable cardioverter defibrillators in children versus adults: Am J Cardiol, 1999; 83(2); 263-66
11.. Eicken A, Kolb C, Lange S, Implantable cardioverter defibrillator (ICD) in children: Int J Cardiol, 2006; 107(1); 30-35
12.. Kies S, Pabelick C, Hurley H, Anesthesia for patients with congenital long QT syndrome: Anesthesiology, 2005; 102(1); 204-10
13.. Tzivoni D, Banai S, Schuger C, Treatment of torsade de pointes with magnesium sulfate: Circulation, 1988; 77(2); 392-97
14.. Thomas S, Behr E, Pharmacological treatment of acquired QT prolongation and torsades de pointes: Br J Clin Pharmacol, 2016; 81(3); 420-27
Figures
In Press
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133