20 May 2021: Articles 
Protein-Activated Kinase 3 (PAK3)-Related Intellectual Disability Associated with Combined Immunodeficiency: A Case Report
Rare coexistence of disease or pathology
Ohood Almutairi1ABDEF*, Hessah A. Almutairi1ABDEF, Maysoun Al Rushood2ADEDOI: 10.12659/AJCR.930966
Am J Case Rep 2021; 22:e930966






Abstract
BACKGROUND: X-linked intellectual disabilities constitute a group of clinically and genetically heterogeneous disorders that are divided into syndromic and nonsyndromic forms. PAK3 mutations are associated with X-linked nonsyndromic forms of intellectual disability, with the most common clinical features being cognitive deficit, large ears, oral motor hypotonia, and neurobehavioral abnormalities. These mutations have been reported to be associated with either loss of the PAK3 protein or loss of its kinase activity. We report a case with the novel PAK3 variant c.685C>T p.(Pro229Ser), which has not been previously described.
CASE REPORT: We report the first case of a PAK3 mutation to present with the common clinical features along with immunodeficiency resembling common variable immune deficiency. Our patient was a 10-year-old girl who had experienced septic shock with a rapidly deteriorating course when she was 5-years-old. The initial immune work-up showed lymphopenia affecting all cell lines, but preferentially the B-cell compartment. Further work-up of this patient revealed low levels of immunoglobulin (Ig) G, undetectable IgA, reduced IgG1 and IgG2 subclasses, and poor response to the diphtheria/tetanus vaccine. Lymphocyte function, tested as the response to the mitogen phytohemagglutinin, was low and fluctuated between 9% and 22% compared with control samples. The patient experienced recurrent respiratory tract infections, and she responded well to regular intravenous Ig treatment and antibiotic prophylaxis.
CONCLUSIONS: The current case might provide a new insight into PAK3 gene function. Although further evidence is needed, it is worth considering that immunological abnormalities may be associated with PAK3 gene mutations.
Keywords: case reports, Common Variable Immunodeficiency, Developmental Disabilities, intellectual disability, p21-Activated Kinases, Child, Child, Preschool, Mutation, Phenotype, Protein Kinases
Background
X-linked intellectual disabilities constitute a group of clinically and genetically heterogeneous disorders that are divided into syndromic and nonsyndromic forms [1,2]. The syndromic form refers to cases that have recognizable dysmorphic features and other distinguishing symptoms and signs. To date, the number of genes associated with an X-linked intellectual disability has increased by 96% from 72 to 141 genes [3]. Among these genes is the protein-activated kinase 3 (
PAK3 mRNA is highly expressed in the fetal human brain but is not found at detectable levels in other fetal organs [12]. The
Here, we describe the first reported case presenting with immunodeficiency and X-linked intellectual disability associated with a novel
Case Report
Our patient was a 10-year-old girl whose parents were nonconsanguineous. The parents were healthy, with no intellectual disability, and both were university graduates. The patient had 1 healthy older brother and no significant family history except for a resolved seizure disorder in 1 maternal cousin. The girl was born full term via spontaneous uncomplicated vaginal delivery, and the fetal and early neonatal periods were unremarkable. Birth weight, length, and head circumference were all within the normal limits.
At the age of 2 weeks, she began having feeding difficulties and was admitted to the hospital for pneumonia. Thereafter, the patient had recurrent lower respiratory tract infections that were attributed to severe gastroesophageal reflux disease and recurrent aspiration.
At the age of 2 years, she was evaluated for failure to thrive and was found to have microcephaly and developmental delay. In addition, hypothyroidism and growth hormone deficiency were diagnosed, and she received treatment for these conditions. Brain magnetic resonance imaging (MRI) revealed diffuse cerebral and cerebellar atrophy with thinning of the corpus callosum, while an electroencephalogram was normal.
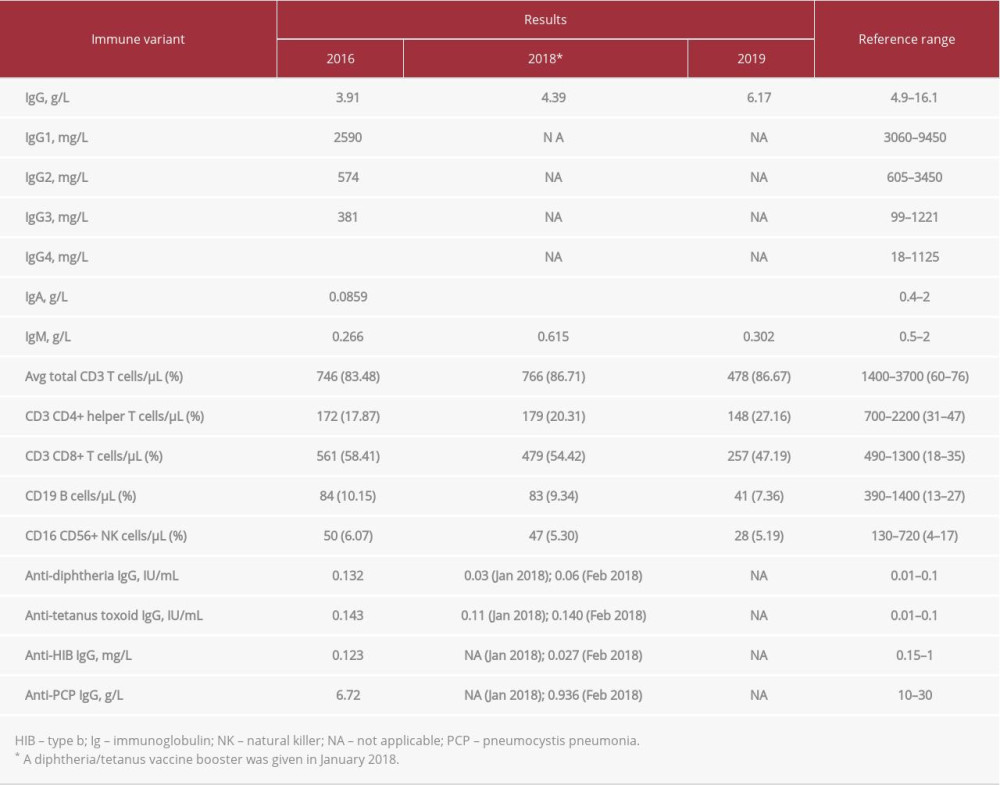
At the age of 5 years, the patient developed septic shock with a rapidly deteriorating course. The initial immune work showed lymphopenia affecting all cell lines, but preferentially the B-cell compartment. The serum immunoglobulin (Ig) levels showed mildly decreased IgG level for age; however, vaccine-specific antibody titers were protective. Over the following 2 years, the patient’s immune status deteriorated clinically and she experienced recurrent respiratory tract infections. Repeated immune work-up revealed low levels of IgG, undetectable IgA, reduced IgG1 and IgG2 subclasses, and poor response to diphtheria/tetanus vaccine. The patient continued to have lymphopenia, mainly affecting the B-cell compartment. Lymphocyte function, tested through response to the mitogen phytohemagglutinin, was low and fluctuated between 9% and 22% compared with control samples. The patient had normal complement levels (C3 and C4). The patient’s work-up since her diagnosis is shown in Table 2. She was started on daily prophylactic sulfamethoxazole-trimethoprim as well as monthly intravenous Ig in 2018. Since then, the patient has improved.
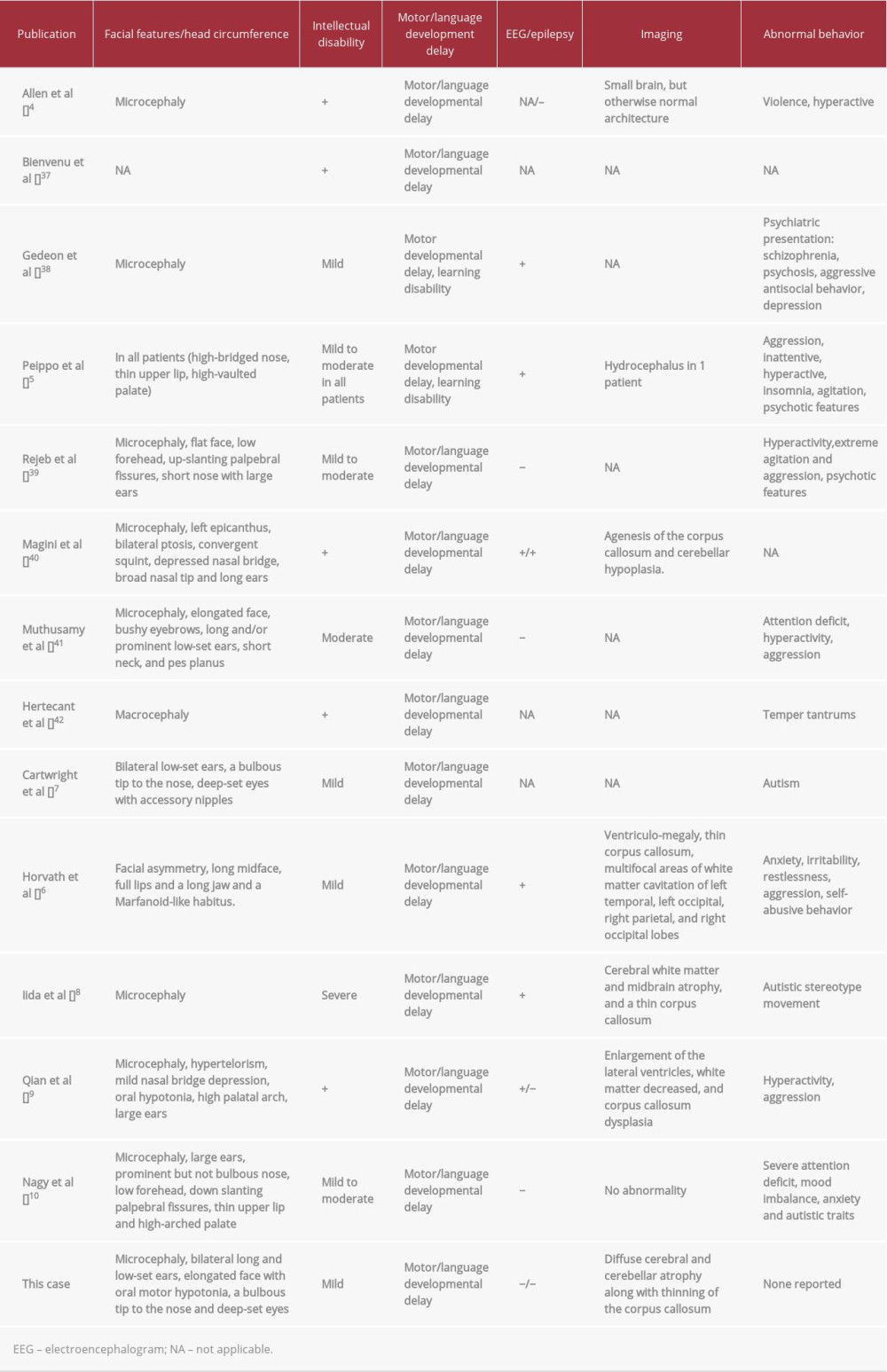
Clinical examination revealed that the patient had microcephaly, bilateral long and low-set ears, an elongated face with oral motor hypotonia, a bulbous tip to the nose, and deep-set eyes. Her weight was just below the 50th percentile and her height was just below the 25th percentile based on age-related norms. In view of her immunodeficiency and dysmorphic features, clinical exome sequencing was carried out. It revealed a hemizygous missense variant in the
This variant is classified as being of uncertain significance by VarSome [15] based on American College of Medical Genetics and Genomics (ACMG) standards and guidelines for interpretation of sequence variants (PM2, PP2, PP3) [16]. This variant is not present in the genome aggregation database nor in the 1000 Genomes Project.
The variant was predicted to be pathogenic by 9 different computational programs (BayesDel addAF, DANN, FATHMM-MKL, LIST-S2, LRT, MutationTaster, PROVEAN, PolyPhen, and PrimateAl). Moreover, it has a genomic evolutionary rate profiling score of 5.8499, with a score of 6.17 being the most conserved.
Paternal carrier testing was recommended to the parents to know whether the variant was due to a de novo or an inherited mutation; however, they were reluctant. The patient was referred for X-chromosome inactivation analysis, but unfortunately, the test could not be done for many reasons.
Discussion
This report describes a case of intellectual disability linked to a mutation in the
This case is also the first to be reported with a
The patient had features similar to those reported for other families carrying a
In a study of
The most recent brain MRI in our patient predictably showed a small atrophied brain with grossly normal brain structures. Similar MRI findings were reported in mentally retarded male patients carrying a
Our patient, similar to others with a
To the best of our knowledge, our report describes the first case of combined immunodeficiency in a patient with this mutation. A relationship between PAK3 mutations and immuno-deficiency has not been previously found, and limited studies have investigated the roles of PAK3 in immunodeficiency. However, the possibility exists that our patient’s intellectual disability and immunodeficiency resulted from 2 different unrelated conditions. Another possibility is that the PAK3 variant could have interacted with another gene to cause the immunodeficiency. This could be supported by the fact that PAK3 can interact with Rac2, a member of the Rho GTPases, which play a pivotal role in regulating several vital cellular and immunological processes [18–20], including lamellipodia formation, chemotaxis, directed migration, and superoxide production in phagocytic cells [21]. Mutations within the encoding region of
In innate immunity, Rho GTPases are involved in phagocytosis and regulation of leukocyte chemotaxis and motility [25–28]. Rho GTPases also play an important role in adaptive immunity by regulating the activation and migration of T and B cells and forming immunological synapses between dendritic cells and antigen-specific T cells, which are prerequisites for inducing an adaptive T-cell response [29–32]. Furthermore, mutations in Rho and Rho-modulating factors have been found to increase the risk of autoimmune diseases and hematopoietic malignancies [33–35].
One study found the expression of PAK1, another member of the PAK family, was detectable in both naive and activated T cells, while PAK3 was evident only in activated T cells [36]. That study also showed that the specific depletion of PAK3 reduces the elongation of activated T cells induced by activation-inducible lymphocyte immuno-mediatory molecule/inducible co-stimulator, a member of the CD28 co-stimulatory receptor family, as efficiently as PAK1. These findings indicated that both PAK1 and PAK3 are independently involved in this immunological process.
Moreover, a member of a family carrying a
Conclusions
In conclusion, our report further confirms that subjects with a
References:
1.. Lubs HA, Stevenson RE, Schwartz CE, Fragile X and X-linked intellectual disability: Four decades of discovery: Am J Hum Genet, 2012; 90(4); 579-90
2.. Ropers HH, Genetics of early onset cognitive impairment: Annu Rev Genomics Hum Genet, 2010; 11; 161-87
3.. Neri G, Schwartz CE, Lubs HA, Stevenson RE, X-linked intellectual disability update 2017: Am J Med Genet A, 2018; 176(6); 1375-88
4.. Allen KM, Gleeson JG, Bagrodia S, PAK3 mutation in nonsyndromic X-linked mental retardation: Nat Genet, 1998; 20(1); 25-30
5.. Peippo M, Koivisto AM, Särkämö T, PAK3 related mental disability: Further characterization of the phenotype: Am J Med Genet A, 2007; 143A(20); 2406-16
6.. Horvath GA, Tarailo-Graovac M, Bartel T, Improvement of self-injury with dopamine and serotonin replacement therapy in a patient with a hemizygous PAK3 mutation: A new therapeutic strategy for neuropsychiatric features of an intellectual disability syndrome: J Child Neurol, 2018; 33(1); 106-13
7.. Cartwright A, Smith K, Balasubramanian M, Short case report: Xq23 deletion involving PAK3 as a novel cause of developmental delay in a 6-year-old boy: Clin Dysmorphol, 2017; 26(1); 38-40
8.. Iida A, Takano K, Takeshita E, A novel PAK3 pathogenic variant identified in two siblings from a Japanese family with X-linked intellectual disability: Case report and review of the literature: Cold Spring Harb Mol Case Stud, 2019; 5(6); a003988
9.. Qian Y, Wu B, Lu Y, Novel PAK3 gene missense variant associated with two Chinese siblings with intellectual disability: A case report: BMC Med Genet, 2020; 21(1); 31
10.. Nagy D, Farkas K, Armengol L, Further delineation of the phenotype of PAK3-associated x-linked intellectual disability: Identification of a novel mis-sense mutation and review of literature: Eur J Med Genet, 2020; 63(4); 103800
11.. Boda B, Alberi S, Nikonenko I, The mental retardation protein PAK3 contributes to synapse formation and plasticity in hippocampus: J Neurosci, 2004; 24(48); 10816-25
12.. Neri G, Chiurazzi P, X-linked mental retardation: Advances in genetics, 1999; 55-94, San Diego, CA, Academic Press
13.. Donnelly AJ, Partington MW, Ryan AK, Mulley JC, Regional localisation of two non-specific X-linked mental retardation genes (MRX30 and MRX31): Am J Med Genet, 1996; 64(1); 113-20
14.. Zhao ZS, Manser E, PAK and other Rho-associated kinases – effectors with surprisingly diverse mechanisms of regulation: Biochem J, 2005; 386(Pt 2); 201-14
15.. Kopanos C, Tsiolkas V, Kouris A, VarSome: The human genomic variant search engine: Bioinformatics, 2019; 35(11); 1978-80
16.. Richards S, Aziz N, Bale S, Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology: Genet Med, 2015; 17(5); 405-24
17.. Huang W, Zhou Z, Asrar S, p21-Activated kinases 1 and 3 control brain size through coordinating neuronal complexity and synaptic properties: Mol Cell Biol, 2011; 31(3); 388-403
18.. , Functional protein associations netwoks October 17, 2020 https://string-db.org/network/8128.ENSONIP00000021368
19.. Mackay DJ, Hall A, Rho GTPases: J Biol Chem, 1998; 273(33); 20685-88
20.. El Masri R, Delon J, RHO GTPases: From new partners to complex immune syndromes: Nat Rev Immunol, 2021 [Online ahead of print]
21.. Scita G, Tenca P, Frittoli E, Signaling from Ras to Rac and beyond: Not just a matter of GEFs: EMBO J, 2000; 19(11); 2393-98
22.. Ambruso DR, Knall C, Abell AN, Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation: Proc Natl Acad Sci USA, 2000; 97(9); 4654-59
23.. Williams DA, Tao W, Yang F, Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency: Blood, 2000; 96(5); 1646-54
24.. Accetta D, Syverson G, Bonacci B, Human phagocyte defect caused by a Rac2 mutation detected by means of neonatal screening for T-cell lymphopenia: J Allergy Clin Immunol, 2011; 127(2); 535-8.e1-2
25.. Bokoch GM, Regulation of innate immunity by Rho GTPases: Trends Cell Biol, 2005; 15(3); 163-71
26.. Sánchez-Madrid F, del Pozo MA, Leukocyte polarization in cell migration and immune interactions: EMBO J, 1999; 18(3); 501-11
27.. Xu J, Wang F, Van Keymeulen A, Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils: Cell, 2003; 114(2); 201-14
28.. Chimini G, Chavrier P, Function of Rho family proteins in actin dynamics during phagocytosis and engulfment: Nat Cell Biol, 2000; 2(10); E191-96
29.. Katakai T, Kondo N, Ueda Y, Kinashi T, Autotaxin produced by stromal cells promotes LFA-1-independent and Rho-dependent interstitial T cell motility in the lymph node paracortex: J Immunol, 2014; 193(2); 617-26
30.. Varga G, Nippe N, Balkow S, LFA-1 contributes to signal I of T-cell activation and to the production of T(h)1 cytokines: J Invest Dermatol, 2010; 130(4); 1005-12
31.. Ocana-Morgner C, Wahren C, Jessberger R, SWAP-70 regulates RhoA/ RhoB-dependent MHCII surface localization in dendritic cells: Blood, 2009; 113(7); 1474-82
32.. Kamon H, Kawabe T, Kitamura H, TRIF-GEFH1-RhoB pathway is involved in MHCII expression on dendritic cells that is critical for CD4 T-cell activation: EMBO J, 2006; 25(17); 4108-19
33.. Rozo C, Chinenov Y, Maharaj RK, Targeting the RhoA-ROCK pathway to reverse T-cell dysfunction in SLE: Ann Rheum Dis, 2017; 76(4); 740-47
34.. Liu Z, Xu Y, Zhang X, The motorized RhoGAP myosin IXb (Myo9b) in leukocytes regulates experimental autoimmune encephalomyelitis induction and recovery: J Neuroimmunol, 2015; 282; 25-32
35.. Cortes JR, Ambesi-Impiombato A, Couronné L, RHOA G17V induces T follicular helper cell specification and promotes lymphomagenesis: Cancer Cell, 2018; 33(2); 259-73.e7
36.. Nukada Y, Okamoto N, Konakahara S, AILIM/ICOS-mediated elongation of activated T cells is regulated by both the PI3-kinase/Akt and Rho family cascade: Int Immunol, 2006; 18(12); 1815-24
37.. Bienvenu T, des Portes V, McDonell N, Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation: Am J Med Genet, 2000; 93(4); 294-98
38.. Gedeon AK, Nelson J, Gécz J, Mulley JC, X-linked mild non-syndromic mental retardation with neuropsychiatric problems and the missense mutation A365E in PAK3: Am J Med Genet A, 2003; 120a(4); 509-17
39.. Rejeb I, Saillour Y, Castelnau L, A novel splice mutation in PAK3 gene underlying mental retardation with neuropsychiatric features: Eur J Hum Genet, 2008; 16(11); 1358-63
40.. Magini P, Pippucci T, Tsai IC, A mutation in PAK3 with a dual molecular effect deregulates the RAS/MAPK pathway and drives an X-linked syndromic phenotype: Hum Mol Genet, 2014; 23(13); 3607-17
41.. Muthusamy B, Selvan LDN, Nguyen TT, Next-generation sequencing reveals novel mutations in X-linked intellectual disability: Omics, 2017; 21(5); 295-303
42.. Hertecant J, Komara M, Nagi A, A de novo mutation in the X-linked PAK3 gene is the underlying cause of intellectual disability and macrocephaly in monozygotic twins: Eur J Med Genet, 2017; 60(4); 212-16
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952658
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953243
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952989
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953049
Most Viewed Current Articles
07 Dec 2021 : Case report
22,697,854
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,914
174,914
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,026
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,962
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133