03 November 2021: Articles 
A Rapidly Progressive Case of Ectopic Adrenocorticotropic Hormone (ACTH) Syndrome
Unusual clinical course, Challenging differential diagnosis, Unusual setting of medical care
Rishi RajDOI: 10.12659/AJCR.934437
Am J Case Rep 2021; 22:e934437






Abstract
BACKGROUND: Adrenocorticotropic hormone (ACTH)-dependent Cushing’s syndrome (CS) secondary to an ectopic source is an uncommon condition, accounting for 4-5% of all cases of CS. Refractory hypokalemia can be the presenting feature in patients with ectopic ACTH syndrome (EAS), and is seen in up to 80% of cases. EAS can be rapidly progressive and life-threatening without timely diagnosis and intervention.
CASE REPORT: We present a case of a 74-year-old White woman who first presented with hypokalemia, refractory to treatment with potassium supplementation and spironolactone. She progressively developed generalized weakness, recurrent falls, bleeding peptic ulcer disease, worsening congestive heart failure, and osteoporotic fracture. A laboratory workup showed hypokalemia, hypernatremia, and primary metabolic alkalosis with respiratory acidosis. Hormonal evaluation showed elevated ACTH, DHEA-S, 24-h urinary free cortisol, and unsuppressed cortisol following an 8 mg dexamethasone suppression test, suggestive of ACTH-dependent CS. CT chest, abdomen, and pelvis, and FDG/PET CT scan showed a 1.4 cm right lung nodule and bilateral adrenal enlargement, confirming the diagnosis of EAS, with a 1.4-cm lung nodule being the likely source of ectopic ACTH secretion. Due to the patient’s advanced age, comorbid conditions, and inability to attend to further evaluation and treatment, her family decided to pursue palliative and hospice care.
CONCLUSIONS: This case illustrates that EAS is a challenging condition and requires a multidisciplinary approach in diagnosis and management, which can be very difficult in resource-limited areas. In addition, a delay in diagnosis and management often results in rapid deterioration of clinical status.
Keywords: Cushing Syndrome, Endocrine System, Hypokalemia, ACTH Syndrome, Ectopic, Adrenocorticotropic Hormone, Female, Humans, Hydrocortisone
Background
Cushing’s syndrome (CS) has a variety of clinical manifestations resulting from excess steroid hormone production from adrenal glands (endogenous) or administration of glucocorticoids (exogenous) [1,2]. Endogenous CS is classified into 2 main categories: ACTH-dependent and ACTH-independent disease. In ACTH-dependent disease, the source of ACTH can further be subdivided into either the pituitary gland or an ectopic source [2]. Ectopic ACTH syndrome (EAS) results from excess production of ACTH from extra-pituitary sources [2] and accounts for approximately 4–5% of cases of CS [3,4]. Common clinical manifestations of CS include weight gain, central obesity, fatigue, plethoric facies, purple striae, hirsutism, irregular menses, hypertension, diabetes/glucose intolerance, anxiety, muscle weakness, bruising, and osteoporosis [2]. Hypokalemia is a less defining feature, seen in roughly 20% of cases with CS. However, it is present in up to 90% of cases with EAS [2,5], which is attributed to the mineralocorticoid action of steroid [6].
Hypercortisolism due to EAS is usually severe and rapid in onset, and excess cortisol levels can lead to severe clinical manifestations, including life-threatening infections [7]. Moreover, in most patients with EAS, the source of excess ACTH is an underlying malignancy that can further result in rapid deterioration of the overall clinical condition. Although numerous malignancies have been associated with EAS, lung neuroendocrine tumors (NETs) are the most common [2,8]. Since the treatment of choice for EAS is complete resection of the tumor, the correct localization of the source of ectopic ACTH is crucial in managing these patients. Traditional radiological investigations can localize these tumors in up to 50% of cases [9]; however, recent studies utilizing somatostatin receptor (SSTR) analogs have increased the sensitivity and specificity of tumor localization [9–11]. This case report describes a challenging case of an elderly patient with EAS who presented with refractory hypokalemia. Her clinical condition deteriorated rapidly in the absence of surgical intervention.
Case Report
A 74-year-old White woman was brought to the Emergency Department from her nephrologist’s office with a chief concern of persistent anasarca and recurrent hypokalemia of 1-month duration. In addition, she reported generalized weakness and recurrent mechanical falls in the preceding 3 months. Before presentation in March 2021, she had a medical history of type 2 diabetes, chronic kidney disease stage 3b, atrial fibrillation on chronic anticoagulation, heart failure with reduced ejection fraction (EF 35–40%), hypothyroidism, hypertension, and hyperlipidemia. Home medications included diltiazem, apixaban, insulin glargine, levothyroxine, simvastatin, carvedilol, glimepiride, sacubitril, valsartan, and furosemide.
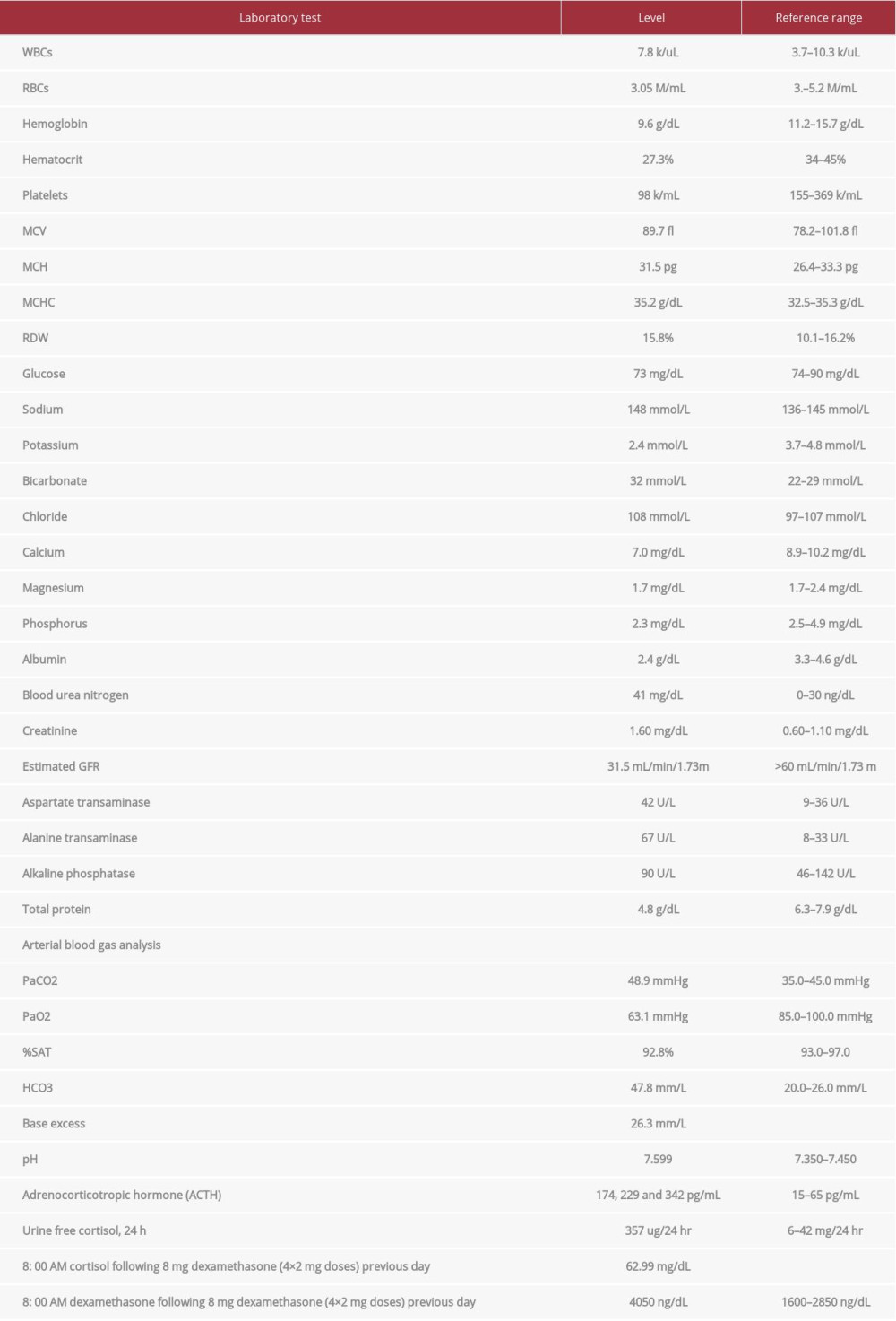
On presentation, she was hemodynamically stable with temperature 36.5°C, heart rate 67 beats per min, blood pressure 139/57 mmHg, respiratory rate 20 per min, and saturation 98% on 2 L oxygen supplementation. Her height was 162.6 cm, and weight was 80.88 kg, with a body mass index (BMI) of 30.6 kg/m2. A physical exam showed central obesity, bruising in extremities, generalized facial swelling mainly in the periorbital region, severe pitting edema in bilateral lower extremities, and moderate pitting edema in bilateral upper extremities. A laboratory workup revealed serum potassium 2.4 mmol/L (3.6–5.2 mmol/L), serum sodium 148 mmol/L (133–144 mmol/L), and eGFR 31.5 mL/min/1.73 m2. Arterial blood gas analysis showed pH 7.6, PaCO2 48.9 mmHg (35.0–45.0 mmHg), and serum bicarbonate 32 mmol/L (22–29 mmol/L), which was consistent with primary metabolic alkalosis, appropriately compensated by respiratory acidosis. Due to concerns of loop diuretic-induced hypokalemia, she was started on spironolactone and potassium replacement. However, potassium levels persistently remained in the low range of 2–3.5 mmol/L (3.6–5.2 mmol/L) despite confirming compliance to medications and adequate up-titration in the dose of spironolactone and potassium chloride. Hence, the workup for the secondary cause of persistent hypokalemia was pursued.
Hormonal evaluation revealed plasma aldosterone concentration (PAC) <1.0 ng/dL, plasma renin activity (PRA) 0.568 ng/mL/h (0.167–5.380 ng/mL/h), 24-h urine free cortisol (UFC) 357 mg/24h (6–42 mg/24h), ACTH 174 pg/mL, and DHEA-S 353 ug/dL (20.4–186.6 ug/dL). ACTH levels on 2 repeat testings were 229 pg/mL and 342 pg/mL. The rest of the laboratory workup is summarized in Table 1. Considering elevated ACTH and 24-h UFC, a preliminary diagnosis of ACTH-dependent Cushing syndrome was made. An 8-mg dexamethasone suppression test revealed non-suppressed cortisol of 62.99 ug/dL along with dexamethasone 4050 ng/dL (1600–2850 ng/dL). A pituitary MRI was unremarkable for any focal lesion suggesting a diagnosis of ACTH-dependent Cushing’s syndrome secondary to an ectopic source. Imaging studies were then performed to determine the source. A CT scan of the chest and abdomen revealed adenomatous thickening with nodularity of bilateral adrenal glands, and a 1.4-cm nodule in the right middle lobe (Figure 1A, 1B). FDG-PET/CT showed severe bilateral enlargement of the adrenal glands with severe hyper-metabolic uptake (mSUV 9.2 and 9.1 for left and right adrenal glands, respectively) (Figure 2A). The uptake of the right lung nodule on PET/CT was 1.4 mSUV (Figure 2B).
Based on unsuppressed cortisol following an 8-mg dexamethasone suppression test, negative pituitary MRI, and 1.4-cm lung nodule, we diagnosed ACTH-dependent CS secondary to an ectopic source, most likely from the 1.4-cm lung nodule. While awaiting localization studies, within 3 months of initial presentation, she had 2 hospitalizations, one in May 2021 for acute anemia secondary to bleeding peptic ulcer disease (PUD) requiring endoscopic clipping of the bleeding ulcer, and another in June 2021 for acute on chronic congestive heart failure. The patient’s overall condition continued to deteriorate, and she became progressively weak and wheelchair-bound. A 68-Ga-DOTATATE was planned to establish the source of ectopic ACTH definitively; however, she developed a left hip fracture in July 2021 and could not present for follow-up care. Therefore, she was started on Mifepristone until curative surgery. However, considering the patient’s advanced comorbid conditions, the increased burden of the patient’s health care needs on her elderly husband, and the inability of other family members to provide necessary healthcare-related support, palliative care was pursued. In August 2021, she developed a sacral decubitus ulcer and community-acquired pneumonia. However, she was still alive while receiving palliative care in a nursing home until September 2021.
Discussion
Ectopic ACTH syndrome (EAS) is defined as secretion of ACTH from an extra-pituitary source and is the cause of Cushing’s syndrome (CS) in approximately 4–5% of cases [3,4]. Clinical features of EAS depend on the rate and amount of ACTH production [12]. Among all forms of Cushing’s (excluding adrenal cortical carcinoma), EAS has the worst outcome, with one of the most extensive combined UK & Athens study demonstrating a 5-year survival rate of 77.6%. Compared to Cushing’s disease (CD), patients with EAS have severe and excessive production of ACTH, resulting in highly elevated cortisol levels. This leads to hypokalemia, metabolic alkalosis, worsening glycemia, hypertension, psychosis, and infections. Metabolic alkalosis and hypokalemia are the 2 most common acid-base and electrolyte abnormalities associated with glucocorticoid excess among these patients. Studies have shown that hypokalemia is seen in up to 90% of patients with EAS. Although hypertension and hypokalemia are often attributed to primary hyperaldosteronism, other causes should be sought. Under normal circumstances, the mineralocorticoid effect of cortisol is insignificant due to local conversion to cortisone by the action of 11 beta-hydroxysteroid dehydrogenase. Excessive cortisol in patients with EAS saturates the action of 11 beta-hydroxysteroid dehydrogenase and leads to the appearance of mineralocorticoid action of cortisol [6]. In our patient, the initial treatment of hypokalemia was unsatisfactory, so additional endocrine workup was pursued. Elevated urinary cortisol excretion, plasma ACTH levels, unsuppressed cortisol following 8 mg dexamethasone, and lung mass on CT scan strongly suggested that the clinical symptoms were due to EAS. Unfortunately, despite diagnosing the underlying condition contributing to the patient’s symptoms, her clinical condition rapidly deteriorated without surgical treatment.
Various factors resulted in delayed diagnosis in our patient. First, the patient sought medical care only 3 months after symptom onset. Second, furosemide, a medication commonly used to treat patients with HFrEF, is a frequent culprit of hypokalemia and often is treated with adequate potassium supplementation. Third, multiple hospitalizations resulted in delays in the proper endocrine workup necessary for establishing hypercortisolism. Fourth, localization of the ectopic source requires advanced imaging studies, which are only available in a few tertiary care centers. Fifth, even after tumor localization with PET/CT scan, there is still a need for a more definitive localization study using Ga-DOTATATE scan, which has a higher specificity. However, it was unavailable in our institution and was only available in a few tertiary care centers, with the nearest center being 2.5 h away. Sixth, the impact of the COVID-19 pandemic also played a critical role in promptly providing critical care necessary to the patient. In addition to those, the social situation of our patient also played an essential role in contributing to delays in diagnosis.
It is well recognized that EAS is associated with various malignancies, mostly of neuroendocrine origin. The most common location of these tumors was found to be the lung (55.3%), followed by the pancreas (8.5%), mediastinum-thymus (7.9%), adrenal glands (6.4%), and gastrointestinal tract (5.4%) [9]. Prompt surgical removal of ectopic ACTH-secreting tumors is the mainstay of therapy in patients with EAS [13]. However, localization of such tumors with conventional therapy is often challenging as the sensitivity to localize the tumor is 50–60% for conventional imaging such as CT, MRI, and FDG-PET [9]. In a study by Isidori et al, nuclear imaging improved the sensitivity of conventional radiological imaging [9]. Moreover, newer imaging technologies using somatostatin receptor (SSTR) analogs such as 68Ga-DOTATATE PET/CT further improve the ability to localize the tumor. 68Ga-DOTATATE PET/CT, approved in 2016 by the Federal Drug Administration (FDA) for imaging well-differentiated NETs, has a high sensitivity (88–93%) and specificity (88–95%) to diagnose carcinoid tumor [14]; however, a systematic review reported a significantly lower sensitivity (76.1%) of 68Ga-DOTATATE PET/CT to diagnose EAS [15].
Once localized, the optimal management of EAS is surgical re-section of the causative tumor, which is often curative. However, until curative surgery is done, patients should be medically managed. Drugs used to reduce cortisol levels include ketoconazole, mitotane, and metyrapone [16, 17]. These are oral medications and decrease cortisol synthesis by inhibiting adrenal enzymes [17]. Etomidate is the only intravenous drug that immediately reduces adrenal steroid production and can be used when acute reduction in cortisol production is desired [16].
Medical management requires frequent monitoring of cortisol levels and titration of dose to achieve low serum and urine cortisol levels. Mifepristone, an anti-progesterone at a higher dose, works as a glucocorticoid receptor antagonist and can be used to block the action of cortisol. Its use results in variable levels of ACTH and cortisol levels in patients with EAS. Hence, hormonal measurement cannot be used to judge therapeutic response, and clinical improvement is the goal of treatment [18]. Drugs inhibiting ACTH secretion by NETs such as kinase inhibitors (vandetanib, sorafenib, or sunitinib) are effective in treating EAS secondary to medullary thyroid cancer [19]. Somatostatin analogs such as octreotide and lanreotide have demonstrated short- and medium-term efficacy in a few EAS patients; however, a few patients failed to improve, necessitating the use of more effective treatment options [19,20]. Hence, they are not considered a first-line drug as monotherapy and should be used in combination with other agents, or as anti-tumoral therapy in non-excisable metastatic well-differentiated NETs [19,20]. Cabergoline, a dopamine agonist, has been used with variable therapeutic effects in a few patients [19]. In 1 patient, the use of combination therapy using Mifepristone and a long-acting octreotide significantly improved EAS [21]. In our patient, we initiated Mifepristone to reduce the burden associated with frequent biochemical monitoring and planned 68Ga-DOTATATE PET/CT to localize the tumor; however, further diagnostic and therapeutic approaches could not be further undertaken per family wishes.
Conclusions
EAS can present with refractory hypokalemia, especially in patients who are already at risk of developing hypokalemia. Diagnosis of EAS is often challenging and requires a multidisciplinary approach. Localization of source of EAS should be done using nuclear imaging, preferably using SSTR analogs, when available. Urgent surgical evaluation remains the mainstay of treatment following tumor localization and can result in a cure. EAS is a rapidly progressive and life-threatening situation that can be fatal if diagnosis or timely intervention is delayed.
Figures
References:
1.. Pluta RM, Burke AE, Golub RM, JAMA patient page. Cushing syndrome and Cushing disease: JAMA, 2011; 306; 2742
2.. Melmed SKR, Rosen C, Auchus R, Goldfine A: Williams textbook of endocrinology, 2020, Elsevier
3.. Rubinstein G, Osswald A, Hoster E, Time to diagnosis in Cushing’s syndrome: A meta-analysis based on 5367 patients: J Clin Endocrinol Metab, 2020; 105; dgz136
4.. Rosset A, Greenman Y, Osher E, Revisiting Cushing syndrome, milder forms are now a common occurrence: A single-center cohort of 76 subjects: Endocr Pract, 2021; 27; 859-65
5.. Fan L, Zhuang Y, Wang Y, Association of hypokalemia with cortisol and ACTH levels in Cushing’s disease: Ann NY Acad Sci, 2020; 1463; 60-66
6.. Jain SH, Sadow PM, Nose V, Dluhy RG, A patient with ectopic cortisol production derived from malignant testicular masses: Nat Clin Pract Endocrinol Metab, 2008; 4; 695-700
7.. Sarlis NJ, Chanock SJ, Nieman LK, Cortisolemic indices predict severe infections in Cushing syndrome due to ectopic production of adrenocorticotropin: J Clin Endocrinol Metab, 2000; 85; 42-47
8.. Isidori AM, Kaltsas GA, Pozza C, The ectopic adrenocorticotropin syndrome: Clinical features, diagnosis, management, and long-term follow-up: J Clin Endocrinol Metab, 2006; 91; 371-77
9.. Isidori AM, Sbardella E, Zatelli MC, Group ABCS, Conventional and nuclear medicine imaging in ectopic Cushing’s syndrome: A systematic review: J Clin Endocrinol Metab, 2015; 100; 3231-44
10.. Righi L, Volante M, Tavaglione V, Somatostatin receptor tissue distribution in lung neuroendocrine tumours: A clinicopathologic and immunohistochemical study of 218 ‘clinically aggressive’ cases: Ann Oncol, 2010; 21; 548-55
11.. Ozkan ZG, Kuyumcu S, Balkose D, The value of somatostatin receptor imaging with In-111 Octreotide and/or Ga-68 DOTATATE in localizing Ectopic ACTH producing tumors: Mol Imaging Radionucl Ther, 2013; 22; 49-55
12.. Paun DL, Vija L, Stan E, Cushing syndrome secondary to ectopic adrenocorticotropic hormone secretion from a Meckel diverticulum neuroendocrine tumor: Aase report: BMC Endocr Disord, 2015; 15; 72
13.. Grigoryan S, Avram AM, Turcu AF, Functional imaging in ectopic Cushing syndrome: Curr Opin Endocrinol Diabetes Obes, 2020; 27; 146-54
14.. Poeppel TD, Binse I, Petersenn S, 68Ga-DOTATOC versus 68Ga-DOTATATE PET/CT in functional imaging of neuroendocrine tumors: J Nucl Med, 2011; 52; 1864-70
15.. Varlamov E, Hinojosa-Amaya JM, Stack M, Fleseriu M, Diagnostic utility of Gallium-68-somatostatin receptor PET/CT in ectopic ACTH-secreting tumors: A systematic literature review and single-center clinical experience: Pituitary, 2019; 22; 445-55
16.. Findling JW, Raff H, Cushing’s syndrome: Important issues in diagnosis and management: J Clin Endocrinol Metab, 2006; 91; 3746-53
17.. Diez JJ, Iglesias P, Pharmacological therapy of Cushing’s syndrome: Drugs and indications: Mini Rev Med Chem, 2007; 7; 467-80
18.. Wannachalee T, Turcu AF, Auchus RJ, Mifepristone in the treatment of the ectopic adrenocorticotropic hormone syndrome: Clin Endocrinol (Oxf), 2018; 89; 570-76
19.. Young J, Haissaguerre M, Viera-Pinto O, Cushing’s syndrome due to ectopic ACTH secretion: An expert operational opinion: Eur J Endocrinol, 2020; 182; R29-58
20.. Pedroncelli AM, Medical treatment of Cushing’s disease: Somatostatin analogues and pasireotide: Neuroendocrinology, 2010; 92(Suppl. 1); 120-24
21.. Moraitis AG, Auchus RJ, Mifepristone improves octreotide efficacy in resistant ectopic Cushing’s syndrome: Case Rep Endocrinol, 2016; 2016; 8453801
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953007
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953581
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952507
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952041
Most Viewed Current Articles
07 Dec 2021 : Case report
22,760,669
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,388
176,388
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,720
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,753
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133