18 May 2022: Articles 
Mutations in and Associated with a Pectus Excavatum Phenotype in 2 Siblings with Osteogenesis Imperfecta
Congenital defects / diseases
Nelimar Cruz-Centeno1ADEF*, Jean F. Saenz-Maisonet2CDEF, Paola M. López-Dones3DEF, Alberto Santiago-Cornier4ABCD, Victor N. Ortiz-Justiniano5ADEDOI: 10.12659/AJCR.935526
Am J Case Rep 2022; 23:e935526






Abstract
BACKGROUND: Osteogenesis imperfecta is a skeletal disease with a range of phenotypes, depending on the genetic mutation. Individuals with osteogenesis imperfecta type I often have mutations in COL1A genes. This disease can be associated with chest wall deformities such as pectus excavatum, but the number of patients with this presentation is limited, and genetic variants associated with this phenotype have not been reported.
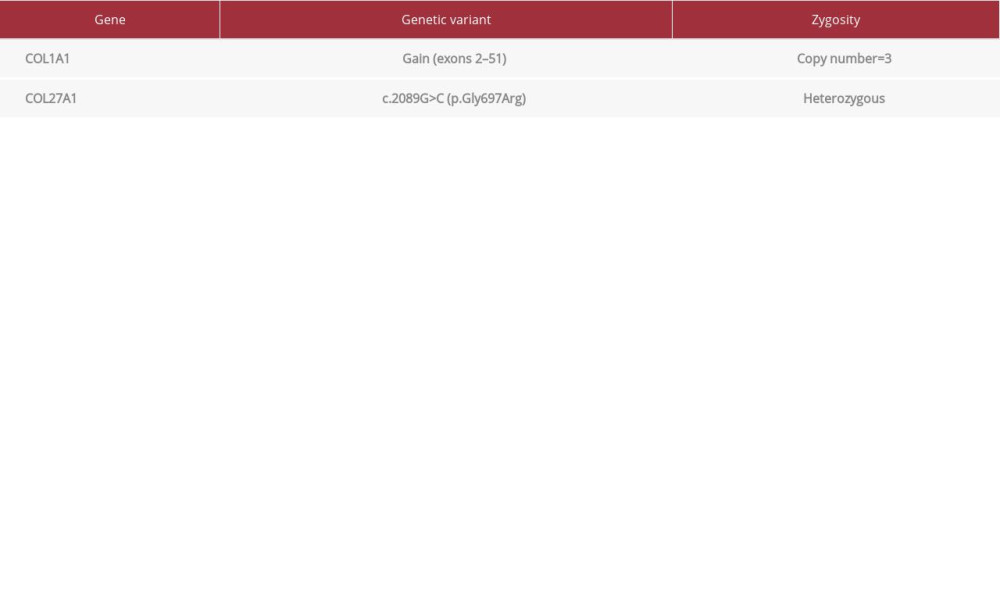
CASE REPORT: We studied the Skeletal Disorders Genetic Panel of 2 siblings with osteogenesis imperfecta type I and severe pectus excavatum requiring surgical correction. Both had severe respiratory symptoms secondary to the chest wall deformity, and the male patient had evidence of mitral valve insufficiency on an echocardiogram. Results of the genetic panel were remarkable for a homozygous copy number gain in exons 2 to 51 in gene COL1A1. Additionally, both had a heterozygous pathogenic variant in exon 7 of gene COL27A1 (replacement of a glycine with arginine in codon 697 of the protein).
CONCLUSIONS: Gene COL27A1 plays a role during the calcification of cartilage to bone and is associated with Steel syndrome, a skeletal disorder mainly found in the Puerto Rican population. Heterozygous carriers of the p.Gly697Arg variant in COL27A1 have not been described to have a phenotype with chest wall deformities. Additionally, a genotype-phenotype relationship regarding pectus excavatum in patients with osteogenesis imperfecta has not been described, suggesting that having COL1A gene mutations and simultaneous haploinsufficiency of COL27A1 can result in a phenotype of osteogenesis imperfecta with pectus excavatum and predispose these patients to additional phenotypic features.
Keywords: Collagen, Funnel Chest, osteogenesis imperfecta, Phenotype, Collagen Type I, Collagen Type I, alpha 1 Chain, Fibrillar Collagens, Humans, Male, Mutation, Siblings
Background
Osteogenesis imperfecta (OI) is a genetic disease most commonly due to a mutation of collagen type I, which is encoded by genes
Steel syndrome, a rare genetic disorder previously known as the “Puerto Rican syndrome,” was first described by Steel et al [4] and is characterized by congenital hip and radial head dislocation, short stature, scoliosis, foot abnormalities, and wrist deformities. In 2015, Gonzaga-Jauregui et al [5] identified the Puerto Rican founder mutation, a homozygous missense variant c.2089G>C (p.Gly697Arg) of the
Case Reports
We report the cases of a 10-year-old boy and 12-year-old girl with autosomal dominant type I OI. The patients were siblings born to a mother who also had OI but no chest wall deformities. The girl started bisphosphonate therapy at approximately 1 year of age, while the boy began therapy at 6 months of age. They are currently receiving intravenous pamidronate therapy every 3 months for 3 consecutive days at a dose of 1 mg/kg.
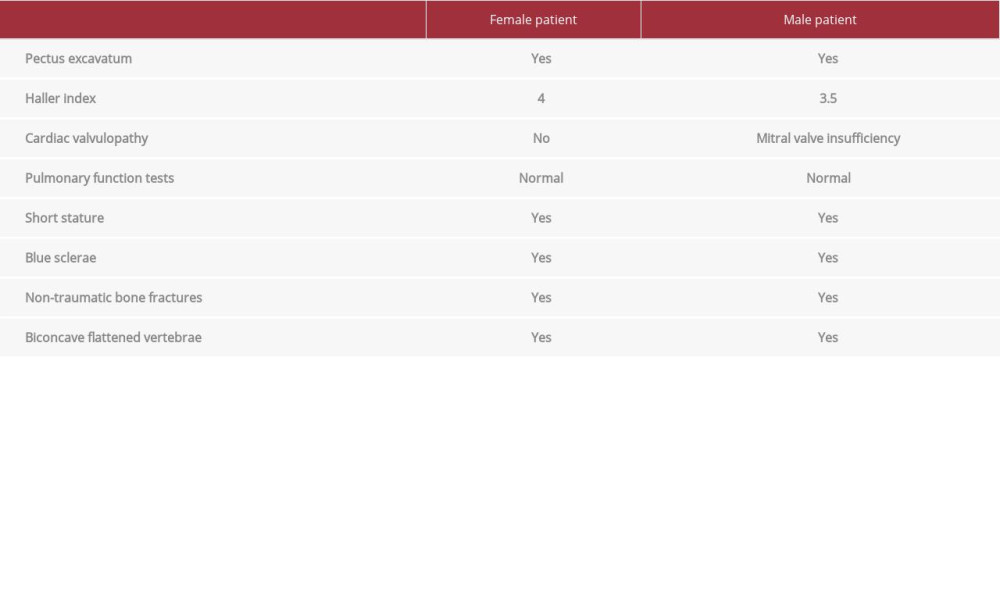
On physical examination, they had severe pectus excavatum, short stature, blue sclerae, history of multiple non-traumatic fractures, and biconcave flattened vertebrae (Table 1). A computed tomography scan of the chest showed a Haller index of 4.0 for the girl and 3.5 for the boy (Figure 1A, 1B). Pulmonary function tests were in the reference range for both patients. Echocardiogram of the boy revealed mitral valve insufficiency; the girl had no cardiac pathology. Both patients were symptomatic with shortness of breath, associated with physical exertion, and limited exercise tolerability. In view of the symptoms, cardiac valvopathy, and Haller index score, both patients met the criteria for surgical correction of the chest wall deformity. The boy underwent the Nuss procedure at 7 years of age and his sister at 8 years. After 3 years of bar placement, they underwent surgical bar removal. Both surgeries were successful, with noticeable improvement in physical activity capacity and correction of the chest wall deformity.
A Skeletal Disorders Panel was conducted to analyze blood samples from both patients (Table 2). Tests were performed following approval of the protocol by an institutional review committee. The results were remarkable for a homozygous copy number gain in exons 2 to 51 in gene
Discussion
Past studies on OI have focused on describing different mutations to the
The
There are several reported mutations in
The
Prior studies have been unable to determine significant relationships between heterozygous carriers of the
It is important to describe this genotype-phenotype relationship because it can guide physicians to recognize genetically predisposed patients and to correct the pectus excavatum early in the clinical course. Pectus excavatum can lead to symptoms such as chest pain, fatigue, dyspnea, respiratory infections, palpitations, and heart murmurs. More serious cardiac concerns include mitral valve prolapse, mitral valve regurgitation, and ventricular compression [12,13]. In patients with skeletal diseases, such as OI, it is known that pectus excavatum increases mortality secondary to cardiac and respiratory problems [13].
Both of our patients had fatigue and intolerance to physical exertion, with a Haller Index >3.25, which is considered severe. The male patient also had evidence of mitral valve insufficiency on echocardiogram, meeting criteria for surgical correction. Identification of the chest wall deformity before puberty was crucial to allow for surgical correction and reversal of pulmonary and cardiac disease. Both patients were asymptomatic at the time of this report, with increased exercise tolerability and improved quality of life. Further genetic testing and molecular studies are in progress, including proteomic studies, to establish the relationship between variants in gene
Conclusions
References:
1.. Lindahl K, Åström E, Rubin CJ, Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteo-genesis imperfecta: Eur J Hum Genet, 2015; 23(8); 1042-50
2.. Cruz-Centeno N, Ortiz-Justiniano VN, Pectus excavatum in osteogenesis imperfecta type I treated with Nuss procedure after pamidronate therapy: J Pediatr Surg Case Rep, 2021; 71; 101915
3.. Zhang DK, Tang JM, Ben XS, Surgical correction of 639 pectus excavatum cases via the Nuss procedure: J Thorac Dis, 2015; 7(9); 1595-605
4.. Steel HH, Piston RW, Clancy M, Betz RR, A syndrome of dislocated hips and radial heads, carpal coalition, and short stature in Puerto Rican children: J Bone Joint Surg Am, 1993; 75(2); 259-64
5.. Gonzaga-Jauregui C, Gamble CN, Yuan B, Mutations in COL27A1 cause Steel syndrome and suggest a founder mutation effect in the Puerto Rican population: Eur J Hum Genet, 2015; 23(3); 342-46
6.. Amlie-Wolf L, Moyer-Harasink S, Carr AM, Three new patients with Steel syndrome and a Puerto Rican specific COL27A1 mutation: Am J Med Genet A, 2020; 182(4); 798-803
7.. Marini JC, Forlino A, Cabral WA, Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans: Hum Mutat, 2007; 28(3); 209-21
8.. Augusciak-Duma A, Witecka J, Sieron AL, Mutations in the COL1A1 and COL1A2 genes associated with osteogenesis imperfecta (OI) types I or III: Acta Biochim Pol, 2018; 65(1); 79-86
9.. Gug C, Caba L, Mozos I, Rare splicing mutation in COL1A1 gene identified by whole exomes sequencing in a patient with osteogenesis imperfecta type I followed by prenatal diagnosis: A case report and review of the literature: Gene, 2020; 741; 144565
10.. Kim JS, Jeon H, Lee H, Biallelic novel mutations of the COL27A1 gene in a patient with Steel syndrome: Hum Genome Var, 2021; 8(1); 17
11.. Gonzaga-Jauregui C, Yesil G, Nistala H, Functional biology of the Steel syndrome founder allele and evidence for clan genomics derivation of COL27A1 pathogenic alleles worldwide: Eur J Hum Genet, 2020; 28(9); 1243-64
12.. Brochhausen C, Turial S, Müller FK, Pectus excavatum: History, hypotheses and treatment options: Interact Cardiovasc Thorac Surg, 2012; 14(6); 801-6
13.. Johnson WR, Fedor D, Singhal S, Systematic review of surgical treatment techniques for adult and pediatric patients with pectus excavatum: J Cardiothorac Surg, 2014; 9; 25
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Most Viewed Current Articles
07 Dec 2021 : Case report  22,759,422
22,759,422
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  175,936
175,936
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,499
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,510
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133