22 August 2022: Articles 
MIRAGE Syndrome Enteropathy Responding to Pancrelipase Despite Normal Pancreatic Fecal Elastase: A Case Report
Challenging differential diagnosis, Unusual or unexpected effect of treatment, Rare disease
Dalwinder Janjua1AEF*, Shiva Shankar1CDEF, Munira AlMaazmi1E, Devendrasing Vijaysing Jadhav1FDOI: 10.12659/AJCR.937057
Am J Case Rep 2022; 23:e937057






Abstract
BACKGROUND: Major findings of myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy (MIRAGE) syndrome is a rare genetic condition caused by a gain-of-function mutation in the SAMD9 gene. It acts as a growth repressor expressed in the endothelial cells. Pathogenic variants in the SAMD9 gene lead to profound growth-restricting activity intrinsic to the protein, which further reduces cellular proliferation and instigates this growth-limiting condition. Gastrointestinal features include chronic diarrhea, severe diaper rash, and colonic dilatation. Until now, there has been no description of exocrine pancreatic insufficiency as a possible cause of enteropathy in MIRAGE syndrome.
CASE REPORT: We report a case of MIRAGE syndrome affecting multiple systems in an infant who had severe enteropathy which responded well to porcine-derived pancreatic enzyme supplements despite normal pancreatic fecal elastase level. The infant is being followed up by multidisciplinary teams in our outpatient department.
CONCLUSIONS: Porcine-derived pancreatic enzyme is beneficial in enteropathy due to MIRAGE syndrome and is worth considering.
Keywords: diarrhea, SAMD9 Protein, Human, Pancrelipase, MIRAGE, adrenal insufficiency, Animals, endothelial cells, Feces, Humans, Pancreatic Elastase, Swine
Background
MIRAGE syndrome is a genetic disorder recently described in 2016 by Narumi et al. It is a multisystem disorder characterized by myelodysplasia, infections, restricted growth, adrenal hypoplasia, genital phenotypes, and enteropathy. It is caused by a heterozygous gain-of-function mutation in the gene encoding sterile alpha motif domain containing protein 9 (SAMD9) on the long arm of chromosome 7q21.2. The characteristic of this disease is hypoplasia of organs due to disturbances in cell growth. Although the precise mechanism underlying this disease is unclear, endosomal dysfunction was speculated to be a possible mechanism for the cell proliferation defects [1]. The mortality rate is high, and most patients die during childhood. Early diagnosis and timely intervention may improve the outcome [2].
Enteropathy can be quite troublesome and difficult to manage, requiring parenteral nutrition. We describe the clinical presentation and management of a neonate with MIRAGE syndrome and enteropathy who showed a good response with porcine-derived pancreatic enzyme supplements despite normal pancreatic fecal elastase level.
Case Report
The infant was born prematurely at 34+5 weeks of gestation by cesarean section, and had intrauterine growth restriction with a birth weight of 1330 grams. The Apgar scores were 8 at 1 min and 8 at 5 min.
The mother was Gravida 3, Para 1. Routine antenatal screening blood tests were normal. Antenatal scans showed intrauterine growth retardation (IUGR), right hydroureter, and female external genitalia. However, the parents had undergone non-invasive prenatal testing (NIPT), which revealed male genotype.
The infant developed respiratory distress soon after birth, requiring support with CPAP. The infant initially showed improvement with weaning of respiratory support over the next 5 days. However, over the next 3 months, the infant intermittently required respiratory support and developed chronic lung disease with persistent need for oxygen.
Clinical assessment soon after birth showed a slightly prominent clitoris, small vaginal opening, small lumps in the groin suggestive of gonads, and no fusion of the labia. Cystovaginoscopy showed normal appearance of the lower vagina, approximately 4 cm in length, ending blindly with no visible cervix and normal female urethra. An ultrasound scan showed features of morphological uterus and vagina without ovaries. Bilateral inguinal canals showed indeterminate soft tissue which could not be characterized as testes or ovary. Surgical exploration of the groin showed the gonad-like structures were very small nubbins with an appearance of atrophic testes. Biopsy revealed immature undifferentiated germ cells and fibrous tissue.
Initial hormonal evaluation revealed low cortisol (125.6 nmol/L), high estradiol (9.15 Pg/ml), low testosterone (0.721 ng/ml), and high luteinizing hormone (3.36 mIU/ml), while 17 OHP was normal. The infant also had low T4 levels, suggestive of hypothyroidism, requiring oral thyroxine supplements.
By day 9 of life, the infant had developed loose stool, metabolic acidosis, and elevated lactate. There was no hyponatremia or hyperkalemia. In view of dehydration, abnormal genital organs, results of NIPT test, and abnormal hormonal profile, DSD was suspected, and the infant was started on hydrocortisone replacement therapy.
An ultrasound scan of the abdomen showed a right hydronephrotic kidney with renal pelvic diameter measuring 12 mm. The left kidney was small and dystrophic with parenchymal calcification. Renal function remained within normal limits. The left kidney gradually involuted with time, with the right kidney showing compensatory growth.
The infant developed watery diarrhea at the age of 2½ months. The infant was on full enteral feeds until then and was started on parenteral nutrition. Attempts to initiate enteral feeding resulted in water diarrhea and dehydration. Stool analysis revealed osmotic diarrhea (stool Na <20, Cl<20, K 118, and Stook osmolar gap >10 mosm/L). Fecal elastase was normal, at 210. Stool for reducing substance was positive, suggestive of lactose intolerance. There also was severe diaper rash, which was difficult to treat. Use of lactose-free and amino acid-based formula did not result in any improvement. Addition of porcine-derived pancreatic enzyme supplements along with amino acid-based formula resulted in significant improvement in the loose stool.
The infant had difficulty in establishing suck feeding due to poor oropharyngeal coordination, microaspiration and severe gastro-esophageal reflux on barium swallow. Infant remained on nasogastric tube feeding initially and was later changed to PEG tube feeding.
By the age of 4½ months, the infant developed seizures requiring multiple anticonvulsant drugs. A CT scan of the skull showed bilateral subdural hematoma in frontal convexities, adjacent to the posterior falx and over the tentorium. MRI brain showed multiple areas of diffusion restriction involving the cerebellar hemisphere, dentate nucleus, and mid-brain, representing areas of acute-to-subacute infarction/ischemia.
MR angiography and MR venography appeared normal. The infant underwent insertion of a subdural shunt, after which the seizures resolved.
At around the same age, the infant developed exposure keratitis and diffuse punctate erosions over both corneas, requiring lubrication. This was due to reduced lacrimation and blinking.
Multiple courses of antibiotics were administered due to concerns of sepsis. CD19 and CD4 counts were low, but immunoglobulin levels were normal.
Exome-based custom panel genetic testing was performed, and sequence analysis of 207 genes related to the infant’s condition identified the presence of a heterozygous pathogenic variant in the gene SAMD9, which is associated with MIRAGE syndrome (p.Arg1293Trp). Parental testing is underway to determine if this variant is de novo.
The infant was discharged home on oxygen, nasogastric feeding, porcine-derived pancreatic enzymes with amino acid-based milk formula. The infant is being followed up in the outpatient department by multidisciplinary teams.
Discussion
MIRAGE syndrome was described in 2016 by Narumi et al in a series of 11 patients [1]. Mutations in the SAM9D gene located on chromosome 7q21.2 are thought to affect protein involved in growth factor signal transduction, resulting in increased antiproliferative effect and ultimately leading to a multisystem growth restriction disorder. Most of these children do not survive beyond the first few years of life.
No consensus is available for clinical diagnostic criteria for MIRAGE syndrome. The diagnosis of MIRAGE syndrome is defined as: 46 XY individuals with 4 or more core features, or 46XX individuals with 3 or more of the core features – myelodysplasia, recurrent infection, restriction of growth, adrenal hypoplasia, and atypical external genitalia in individuals with 46 XY (hypospadias, microphallus, bifid shawl scrotum, ambiguous genitalia, and complete female genitalia) [3]. The other clinical features include prematurity, chronic lung disease, developmental delay, CNS abnormalities, and dysmorphology [4].
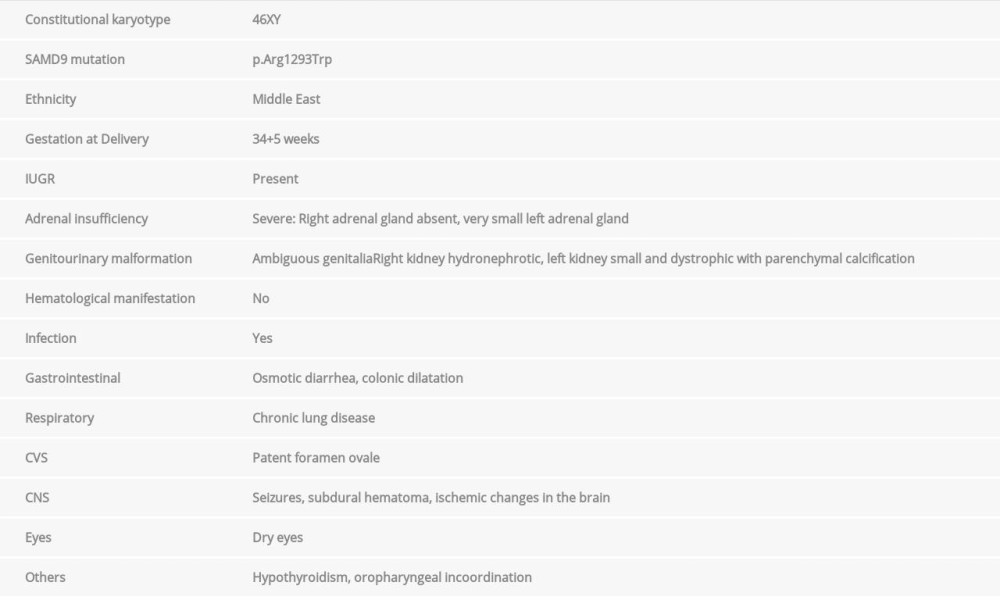
Based on these diagnostic criteria, less than 50 cases have been described so far in the literature. Table 1 shows the characteristics of our patient.
Gastrointestinal complications include chronic diarrhea, aspiration pneumonia, achalasia cardia, and colonic dilatation. Chronic diarrhea occurs in about 80% of the affected individuals and often results in severe diaper rash [3].
The diarrhea can be secretary or osmotic in nature. It was osmotic in nature in our case, as evidenced by stool electrolyte and osmolar gap. Autoimmune enteritis has been thought to be a possible causative factor, and some response to immunosuppression has been described in the literature. An elemental diet has been recommended as a treatment option [5].
Our patient did not respond to amino acid-based fully-hydro-lyzed, lactose-free formula, but did respond to addition of porcine-derived pancreatic enzymes. To date, exocrine pancreatic insufficiency has not been described as a possible cause of diarrhea in MIRAGE syndrome. Fecal elastase levels were normal in our patient.
Fecal elastase 1 level is considered to be the most sensitive and specific indirect test of pancreatic function. Fecal elastase-1 is an enzymatic product of pancreatic secretion that remains relatively stable during transit through the gastrointestinal tract, and there is a direct correlation in pancreatic elastase-1 concentrations in pancreatic fluid and stool.
Despite the fact that fecal elastase-1 levels were normal, our patient responded very well to the addition of porcine-derived pancreatic enzymes. This suggests the possibility of functional deficiency compensated by exogenous pancreatic enzyme supplementation. Further studies may be necessary to clearly establish the impact of pancreatic function on enteropathy in MIRAGE syndrome.
Due to adrenal insufficiency, electrolyte imbalances, myelodysplasia, and predisposition to infections, early recognition may be critical to survival [6,7]. Due to the different genetic etiologies and overlapping clinical features, it is often difficult to make an accurate diagnosis of syndromic adrenal hypoplasia. SAMD9 expression is highest in the fetal adrenal gland. In the study by Narumi et al [1], all evaluated patients had adrenal hypoplasia.
Evaluation of adrenal function is considered very early in the presence of genital abnormalities. Our patient presented with nearly normal-appearing female genitalia. NIPT testing was extremely useful in our case, as the suspicion was heightened because of the difference between the phenotype and the results of NIPT test, and this led us to perform endocrine evaluation and identify a severe cortisol deficiency. The early, pro-active identification of adrenal insufficiency enabled us to initiate replacement therapy early and prevent the ill effects that babies sometimes go through such as shock, hyponatremia, and hyperkalemia.
SAMD9 plays an important role in the development of several body systems. It acts as a growth repressor expressed in the endothelial cells in a wide variety of tissues and to a lesser extent in fibroblasts. Pathogenic variants in the SAMD9 gene lead to profound growth-restricting activity intrinsic to the protein, which further reduces cellular proliferation and instigates this growth-limiting condition [8].
Early interventions such as physiotherapy and occupational therapy maximize developmental potential. The comprehensive care plan should include sub-specialties like genetics, endocrinology, immunology, hemato-oncology, urology, gastroenterology, pulmonology, pediatric surgery, and developmental and behavioral pediatrics. Due to its uniqueness, several questions remain unsolved. Further studies are warranted to reveal the genetics involved in this newly diagnosed syndrome.
Conclusions
In this report, we highlight the usefulness of NIPT in early detection and management of serious complications relating to DSD. Porcine-derived pancreatic enzymes are worth considering in enteropathy due to MIRAGE syndrome, even if the investigations show normal pancreatic functions.
References:
1.. Narumi S, Amano N, Ishii T, SAMD9 mutations cause a novel multi-system disorder, MIRAGE syndrome, and are associated with loss of chromosome7: Nat Genet, 2016; 48(7); 792-99
2.. Shima H, Koehler K, Nomura Y, Role of somatic second-site reversion SAMD9 mutations: J Med Genet, 2018; 55; 81-85
3.. Tanase-Nakao K, Olson TS, Adam MP, Ardinger HH, Pagon RA: GeneReviews® [Internet]; 1993-2022, Seattle (WA), University of Washington Seattle
4.. Jeffries L, Shima H, Ji W, A novel SAMD9 mutation causing MIRAGE syndrome: an expansion and review of phenotype, dysmorphology, and natural history: Am J Med Genet, 2018; 176A; 415-20
5.. Chen B, Avinashi V, SAMD9 associated enteropathy in MIRAGE syndrome: Poster 207. Proceedings of the Clinical Vignette Abstracts NASPGHAN Nov 2, 2017, Las Vegas, NV
6.. Parker TM, Klaassen RJ, Johnston DL, Spontaneous remission of myelodys-plastic syndrome with monosomy 7in a young boy: Cancer Genet Cytogenet, 2008; 182(2); 122-25
7.. Leung EW, Woodman RC, Roland B, Transient myelodysplastic syndrome associated with isochromosome 7q abnormality: Pediatr Hematol Oncol, 2003; 20(7); 539-45
8.. Buonocore F, Kuhnen P, Suntharalingham JP, Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans: J Clin Invest, 2017; 127(5); 1700-13
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952791

Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Most Viewed Current Articles
07 Dec 2021 : Case report
22,759,422
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  175,936
175,936
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,499
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,510
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133