13 April 2023: Articles 
Severe Hemolytic Anemia and Metabolic Acidosis at Birth with Glutathione Synthetase Deficiency and Progressive Neurological Symptoms on Follow-Up
Rare disease
Satoshi Ekuni1ABCDEF, Kei Hirayama2ABCDEF, Miwako Nagasaka1ADEF*, Keita OsumiDOI: 10.12659/AJCR.938396
Am J Case Rep 2023; 24:e938396






Abstract
BACKGROUND: Glutathione synthetase deficiency (GSD) is a rare autosomal recessive disorder caused by glutathione synthetase (GSS) gene variants that occur in 1 in 1 million individuals. The severe form of GSD is characterized by hemolytic anemia, metabolic acidosis with 5-oxoprolinuria, progressive neurological symptoms, and recurrent bacterial infections. This case report presents a male Japanese infant with severe hemolytic anemia and metabolic acidosis at birth caused by GSD, who developed progressive neurological symptoms on follow-up.
CASE REPORT: A Japanese male term infant developed severe hemolytic anemia and metabolic acidosis in the early neonatal period. We suspected GSD based on his symptoms and a high 5-oxoproline urine concentration. We began correcting his metabolic acidosis and administering vitamins C and E supplements. The patient required blood transfusion twice during the acute phase for hemolytic anemia. After age 1 month, he maintained good control of metabolic acidosis and hemolytic anemia. A definitive diagnosis of GSD was made based on high concentrations of 5-oxoproline in urine, low concentrations of glutathione and GSS activity in erythrocytes, and genetic testing. Several episodes of febrile convulsions were started at age 11 months, but none occurred after 2 years. At the last follow-up at age 25 months, metabolic acidosis and hemolytic anemia were well controlled, but he had mild neurodevelopmental delay.
CONCLUSIONS: This case report shows that GSD can present with severe hemolytic anemia and metabolic acidosis at birth, and manifest with subsequent neurological impairment despite early diagnosis and treatment. Therefore, a careful long-term follow-up that includes neurological evaluation is essential for patients with GSD.
Keywords: Acidosis, Anemia, Hemolytic, glutathione synthetase deficiency, Jaundice, Neonatal, Metabolic Diseases, Infant, Newborn, Infant, Humans, Male, Child, Preschool, Glutathione Synthase, Pyrrolidonecarboxylic Acid, Follow-Up Studies
Background
Glutathione synthetase deficiency (GSD) is a rare autosomal recessive disorder caused by mutations in the
Patients with GSD present with hemolytic anemia alone or with metabolic acidosis, neurological symptoms, and recurrent bacterial infections. To date, more than 80 cases in 50 families have been reported worldwide [2–4]. A classification based on the severity of clinical symptoms has been proposed: mild, moderate, and severe GSD [5]. Patients with mild GSD have hemolytic anemia as their only clinical symptom. Those with moderate GSD typically show the symptoms in the neonatal period, including hemolytic anemia, metabolic acidosis, and 5-oxoprolinuria. Those with severe GSD develop progressive neurological symptoms and, occasionally, recurrent bacterial infections, probably due to defective granulocyte function [5]. Ristoff et al reported that 31% (5/16) of patients with severe GSD died before the age of 1 year [5]. In addition, Ameur et al reported a patient with GSD who died from severe bacterial infection and metabolic crisis at age 15 months [6]. The severe form of GSD is a life-threatening disease in infants.
The diagnosis of GSD is made by elevated urinary 5-oxypro-line by gas chromatography-mass spectrometry, decreased GSS activity on red blood cells, and sequencing analysis of the
Currently, no fundamental treatments exist for GSD; therefore, prognosis depends on the quality of supportive care. Metabolic acidosis can be corrected by administering sodium bicarbonate or citric acid, and the dose should be increased to reduce the risk of acute exacerbation, especially in patients with acute infections. Reduced GSH concentrations increase the vulnerability of various tissues to oxidative stress; thus, supplementation with vitamins C (100 mg/kg/day) and E (10 mg/kg/day) is recommended to compensate for the loss of free radical scavenging and to improve the neurological prognosis [7,8]. In fact, Atwal et al reported a case of GSD with good intellectual development after 19 years of continuous vitamin therapy [8]. Vitamin E improves granulocyte function and normalizes microtubule assembly in phagocytosis [5,9].
This case report presents a male Japanese infant with severe hemolytic anemia and metabolic acidosis at birth due to GSD, who developed progressive neurological symptoms on follow-up.
Case Report
A Japanese male infant was born at 38 weeks and 0 days of gestational age to nonconsanguineous Japanese parents with unremarkable medical history. The patient’s father and older brother had recurrent febrile convulsions in infancy. Fetal growth restriction was noted at 24 weeks of gestation. He was born via induced delivery because the reversal of fetal umbilical artery was noted at 38 weeks of gestation. Apgar scores were 10 and 10 at 1 and 5 min, respectively. At birth, the weight was 2174 g (−2.2 standard deviation [SD]), length was 44.5 cm (−1.7 SD), and head circumference was 32.0 cm (−0.7 SD), without marked abnormal physical presentation. Umbilical cord blood gas analyses showed the following: pH, 7.249 (normal range 7.15–7.38); pCO2, 45.0 (normal range 32–68) mmHg; HCO3–, 19.7 (normal range 15.4–26.8) mmol/L; and hemoglobin, 10.4 (normal range 12.5–15.6 at 38 weeks of gestation by fetal blood sampling) g/dL. He was orally fed on breast milk and developed neonatal jaundice at 2 days of age. However, he gradually developed difficulty in breathing and oral feeding. At 5 days of age, venous blood gas analysis and blood tests revealed anemia (hemoglobin, 8.3 [normal range 15.3–18.7] g/dL) and severe metabolic acidosis without lactic acidosis (pH, 6.84 [normal range 7.35–7.45]; HCO3–, 2.9 [normal range 24–26] mmol/L.; base excess, −29.5 [normal range±2] mmol/L; lactate, 11.7 mg/dL [normal range <20] mg/dL; total ketone bodies, 1,275 [normal range <130] μmol/L). Respiratory distress worsened, and mechanical ventilation was required for 2 days.
He was diagnosed with hemolytic anemia based on the elevated concentration of indirect bilirubin (24.1 mg/dL) and the presence of schistocytes in the peripheral blood smear. He was subsequently administered intravenous sodium bicarbonate and erythrocyte transfusion. Urine organic acid analysis revealed a high 5-oxoproline concentration in the urine (5668 [normal range, 27.1–79.3] µg/mg Cr); therefore, GSD was suspected, and he was treated with a vitamin cocktail containing vitamins C and E. The initial improvement in metabolic acidosis worsened the next day and the patient required daily intravenous sodium bicarbonate. Similarly, the hemoglobin concentration initially increased followed by a gradual decline. After the acute phase, he could feed on breast milk while on regular oral treatment with sodium bicarbonate, vitamin C (100 mg/kg/day), and vitamin E (10 mg/kg/day) (Figure 1).
He was discharged at 47 days of age. Since his last red blood cell transfusion at 39 days of age, he has had chronic anemia (7.8–9.6 g/dL) due to GSD but has not had hemolytic attacks requiring red blood cell transfusion. Although subcutaneous erythropoietin administration was initiated on day 66, it had no apparent effect and was terminated on day 105.
He had difficulty taking sodium bicarbonate orally; therefore, we switched to citric acid at 2 months of age.
A subsequent GSH assay in erythrocytes revealed decreased concentrations of GSH (10 [normal range, 62.4–98.9] mg/dL red blood cells) and GSS activity (0.18 [normal range, 0.40–0.78] U/g hemoglobin). Target capture sequencing analysis for congenital hemolytic anemia-related genes, including the 68 genes, was performed [10,11]. Written informed consent for genetic studies was obtained from the parents and the report was approved by the Human Ethics Committee of Tokyo Women’s Medical University. Novel compound heterozygous variants of the
At 11 months, he was admitted for clustered and prolonged febrile convulsions. Diazepam and fosphenytoin sodium hydrate had no effect on the convulsions; however, convulsions did not occur after phenobarbital administration (20 mg/kg, single injection). Electroencephalography revealed normal findings. Brain magnetic resonance imaging (MRI) showed mild ventricular enlargement and corpus callosum hypoplasia. He was discharged 3 days later.
The patient was subsequently hospitalized several times for febrile convulsions, viral infection, and asthma. Nevertheless, he had only 2 episodes of metabolic acidosis: gastroenteritis and viral pneumonia at 1 year of age. His metabolic acidosis was usually well controlled. However, he subsequently experienced recurrent febrile convulsions again, and oral administration of valproate sodium was initiated. After age 2 years, he has been stable without recurring seizures.
Developmental delays became apparent during infancy. He could sit without support at 9 months, stand while holding on to something at 12 months, began to walk at 20 months, and spoke a few words at 24 months. The Kyoto Scale of Psychological Development test [12] was performed at 19 months. His developmental quotient was 63, indicating mild developmental delay. Brain MRI at 25 months showed dilated ventricular and mild brain atrophy (Figure 2).
Discussion
In this case, we suspected GSD relatively early and started acidosis correction and vitamin administration; however, mild developmental delay in infancy was observed at follow-up. Since neurological symptoms may present even after infancy, ongoing long-term medical care and support are required.
GSD is an inherited metabolic disease with reduced GSS activity and low concentrations of GSH; it follows an autosomal recessive inheritance, and is caused by a variety of genetic alterations in the
Additionally, the decrease in GSH concentration in erythrocytes leads to oxidation and degeneration of the erythrocyte membrane, leading to hemolysis, which causes hemolytic anemia and jaundice. A decline in hemolysis in adulthood can be explained by a decrease in lipid peroxidation in the erythrocyte membrane or an increase in hydrogen peroxide detoxification with increasing age [14]. In patients with GSD, neurological symptoms are considered to result from several factors, including exposure to oxidative stress induced by the accumulating free radicals, as well as altered concentrations of GSH, which is also an important neuromodulator and neurotransmitter in the central nervous system [9]. Additionally, kernicterus caused as a result of hemolysis can cause neurological symptoms. The present case showed no signs or symptoms of athetosis, and brain MRI did not show any abnormalities in the basal ganglia, indicating that the mild developmental delay observed in the present case was not associated with severe jaundice in the neonatal period. Based on the clinical course, EEG findings, and family history, we have determined that the patient has febrile seizures, but there is a high possibility that the seizures may progress to epilepsy, so the patient will be monitored carefully.
Patients with GSD reportedly present with increased susceptibility to infection derived from defective granulocyte function [5,15]. Njålsson et al reported that mortality in the neonatal period was attributable to infection or electrolyte abnormalities in 25% of the patients with GSD [15]. In addition, Ameur et al reported a case of GSD that resulted in death at age 15 months from bacterial septic shock and severe metabolic acidosis [6]. In our case, the patient was hospitalized several times for viral infections, but his general condition was not severe. In patients with GSD, it is important to take early action during infection.
Some reviews in patients with GSD have stated that it is best to avoid administration of drugs that can induce hemolytic crises in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency, such as phenobarbital and acetylsalicylic acid [5,16], but phenobarbital is not a contraindicated drug for G6PD deficiency [17]. Also in our case, high-dose administration of phenobarbital did not cause any adverse effects. Single-dose phenobarbital may be used without hemolytic crises in patients with GSD.
The current patient harbored novel compound heterozygous variants (c. 430G>A: p. Glu144Lys, c. 805G>A: p. Gly269Ser) inherited from his parents. These residues are highly conserved between species (Figure 4A) [18]. The Glu144 residue is in magnesium binding site and ATP binding site, and the Gly269 residue is in substrate biding site (Figure 4B) [19]. Several pathogenic variants (p. Arg267Trp, p. Tyr270Cys, and p. Tyr270His) were previously reported in the vicinity of the Gly269 residue [7]. These variants (p. Glu144Lys and p. Gly269Ser) were predicted to be disease-causing by several major
In a study of 16 severely affected patients with metabolic acidosis, hemolytic anemia, and neurological symptoms, 7 of them died, 5 of which died before the age of 1 year [5]. Hence, the severe form of GSD is a life-threatening disease in infants. Ristoff et al indicated that early diagnosis, acidosis correction, and early supplementation with vitamins C and E were the most important determinants of favorable long-term clinical outcomes in patients with GSD [5].
In the current case, the treatment was initiated at 5 days of age and the presumptive diagnosis of GSD was reached based on high urine concentrations of 5-oxoproline determined with urine organic acid analysis conducted at 11 days of age. He was diagnosed and started treatment relatively early, which saved his life, but he has developmental delay with a severe form of GSD. Even when metabolic acidosis and hemolytic anemia are well controlled, neurologic findings, such as seizures and developmental delays, may present after infancy.
However, other clinical courses may also occur. Atwal et al reported a case of severe GSD treated with long-term vitamin therapy and acidosis correction with follow-up until 19 years. The GSD initially manifested with developmental delay in early childhood, but the patient later had normal intellectual development [8]. The neurodevelopmental course may be variable, and we believe that it is important to continue vitamin therapy and correction of metabolic acidosis with long-term follow-up.
Conclusions
As shown in this case report, GSD can present with severe hemolytic anemia and metabolic acidosis at birth and manifest with subsequent neurological impairment in infancy. Early diagnosis, starting treatment as soon as possible and continuing medication, and long-term follow-up that includes neurological evaluation are essential for patient with GSD.
Figures
References:
1.. Webb GC, Vaska VL, Gali RR, The gene encoding human glutathione synthetase (GSS) maps to the long arm of chromosome 20 at band 11.2: Genomics, 1995; 30(3); 617-19
2.. Ristoff E, Larsson A, Inborn errors in the metabolism of glutathione: Orphanet J Rare Dis, 2007; 2; 16
3.. Iyori H, Hirono A, Kobayashi N, Glutathione synthetase deficiency: Rinsho Ketsueki, 1996; 37; 329-34
4.. Xia H, Ye J, Wang L, A case of severe glutathione synthetase deficiency with novel GSS mutations: Braz J Med Biol Res, 2018; 51; e6853
5.. Ristoff E, Mayatepek E, Larsson A, Long-term clinical outcome in patients with glutathione synthetase deficiency: J Pediatr, 2001; 139; 79-84
6.. Ben Ameur S, Aloulou H, Nasrallah F, Hemolytic anemia and metabolic acidosis: Think about glutathione synthetase deficiency: Fetal Pediatr Pathol, 2015; 34; 18-20
7.. Njålsson R, Ristoff E, Carlsson K, Genotype, enzyme activity, glutathione level, and clinical phenotype in patients with glutathione synthetase deficiency: Hum Genet, 2005; 116; 384-89
8.. Atwal PS, Medina CR, Burrage LC, Sutton VR, Nineteen-year follow-up of a patient with severe glutathione synthetase deficiency: J Hum Genet, 2016; 61; 669-72
9.. Janáky R, Ogita K, Pasqualotto BA, Glutathione and signal transduction in the mammalian CNS: J Neurochem, 1999; 73; 889-902
10.. Kanno H, Ogura H, [Diagnosis of congenital hemolytic anemia by comprehensive gene analysis: significance and limitations.]: Rinsho Ketsueki, 2021; 62; 472-79 [in Japanese]
11.. Yamamoto KS, Utshigisawa T, Kanno H, Clinical and genetic diagnosis of thirteen Japanese patients with hereditary spherocytosis: H Hum Genome Var, 2022; 9; 1
12.. Aoki S, Hashimoto K, Ikeda N, Comparison of the Kyoto scale of psychological Development 2001 with the parent-rated Kinder Infant Development Scale (KIDS): Brain Dev, 2016; 38; 481-90
13.. Guney Varal I, Dogan P, Glutathione synthetase deficiency: A novel mutation with femur agenesis: Fetal Pediatr Pathol, 2020; 39; 38-44
14.. Younkin S, Oski FA, Barness LA, Mechanism of the hydrogen peroxide hemolysis test and its reversal with phenols: Am J Clin Nutr, 1971; 24; 7-13
15.. Njålsson R, Glutathione synthetase deficiency: Cell Mol Life Sci, 2005; 62; 1938-45
16.. Larsson A, Anderson ME, Glutathione synthetase deficiency and other disorders of the gamma-glutamyl cycle: The metabolic and molecular bases of inherited disease, 2001; 2205-16, New York, McGraw Hill Education
17.. Beulter E, G6PD Deficiency: Blood, 1994; 84; 3613-36
18.. , HomoloGene accessed date 2/17/2023 https://www.ncbi.nlm.nih.gov/homologene?cmd=Retrieve&dopt=MultipleAlignment&list_uids=148
19.. accessed date 8/26/2022. https://www.uniprot.org/uniprot/P48637
Figures
Tables
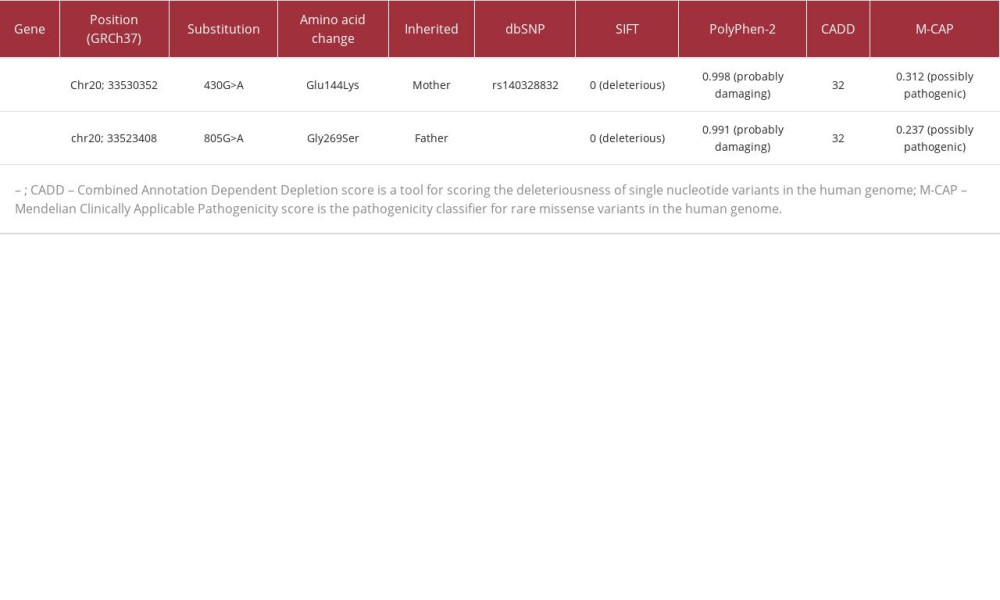 Table 1.. Results of in silico predictive algorithms for variants in the present case. Multiple lines of computational analysis showed supportive data of deleterious effects on the gene or gene product.Table 1.. Results of in silico predictive algorithms for variants in the present case. Multiple lines of computational analysis showed supportive data of deleterious effects on the gene or gene product.
Table 1.. Results of in silico predictive algorithms for variants in the present case. Multiple lines of computational analysis showed supportive data of deleterious effects on the gene or gene product.Table 1.. Results of in silico predictive algorithms for variants in the present case. Multiple lines of computational analysis showed supportive data of deleterious effects on the gene or gene product. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133