27 April 2023: Articles 
Case Series of Genetically Confirmed Index Cases of Familial Hypercholesterolemia in Primary Care
Unusual setting of medical care, Rare disease
Aisyah Kamal1BCDEF, Johanes Dedi Kanchau1BCDEF, Nur Syahirah Shahuri1BCDE, Mohamed-Syarif Mohamed-Yassin2DE, Noorhida Baharudin12DE, Suraya Abdul Razak123DE, Siti Fatimah Badlishah-Sham2DE, Hasidah Abdul-Hamid24DE, Aznida Firzah Abdul Aziz5DE, Alyaa Al-Khateeb16DEF, Yung An Chua16DEF, Noor Alicezah Mohd Kasim17DE, Siti Hamimah Sheikh Abdul Kadir89DEF, Hapizah Nawawi1DE, Nadeem Qureshi4DE, Anis Safura Ramli12ADEG*DOI: 10.12659/AJCR.939489
Am J Case Rep 2023; 24:e939489






Abstract
BACKGROUND: In Malaysia, the prevalence of genetically confirmed heterozygous familial hypercholesterolemia (FH) was reported as 1 in 427. Despite this, FH remains largely underdiagnosed and undertreated in primary care.
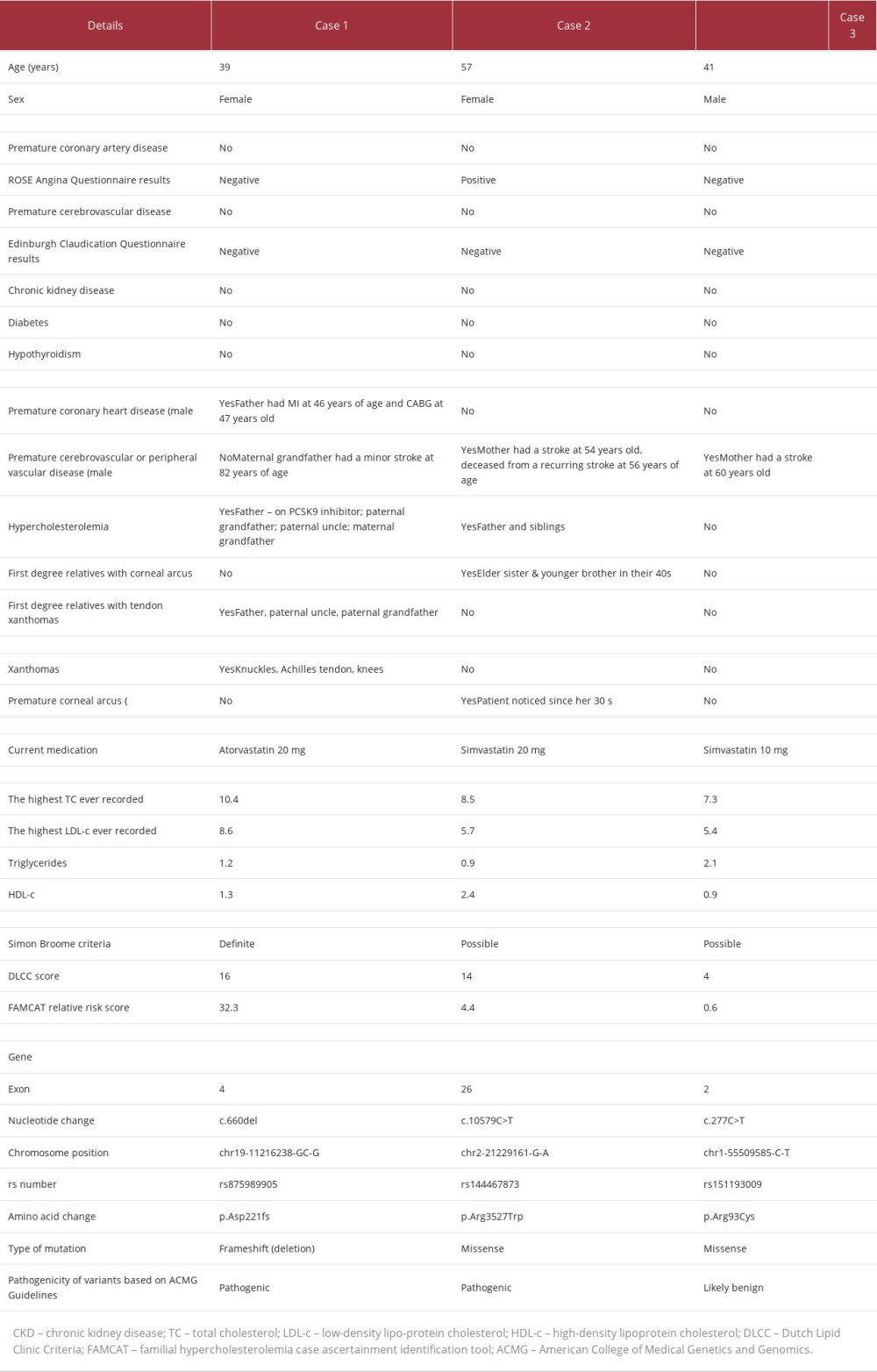
CASE REPORT: In this case series, we report 3 FH cases detected in primary care due to mutations in the low-density lipoprotein receptor (LDLR), apolipoprotein-B (APOB), and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes. The mutations in case 1 (frameshift c.660del pathogenic variant in LDLR gene) and case 2 (missense c.10579C>T pathogenic variant in APOB gene) were confirmed as pathogenic, while the mutation in case 3 (missense c.277C>T mutation in PCSK9 gene) may have been benign. In case 1, the patient had the highest LDL-c level, 8.6 mmol/L, and prominent tendon xanthomas. In case 2, the patient had an LDL-c level of 5.7 mmol/L and premature corneal arcus. In case 3, the patient had an LDL-c level of 5.4 mmol/L but had neither of the classical physical findings. Genetic counseling and diagnosis were delivered by primary care physicians. These index cases were initially managed in primary care with statins and therapeutic lifestyle modifications. They were referred to the lipid specialists for up-titration of lipid lowering medications. First-degree relatives were identified and referred for cascade testing.
CONCLUSIONS: This case series highlights different phenotypical expressions in patients with 3 different FH genetic mutations. Primary care physicians should play a pivotal role in the detection of FH index cases, genetic testing, management, and cascade screening of family members, in partnership with lipid specialists.
Keywords: atherosclerosis, Coronary Artery Disease, Hyperlipidemia with Familial Hypercholesterolemic Xanthomatosis, Physicians, Primary Care, Humans, Proprotein Convertase 9, Cholesterol, LDL, Phenotype, Hyperlipoproteinemia Type II, Mutation, Apolipoproteins B, Primary Health Care
Background
Familial hypercholesterolemia (FH) is an inherited genetic disorder with an autosomal dominant mode of inheritance [1]. It is one of the most common monogenic diseases that leads to an elevated risk of premature atherosclerosis, due to its effect on plasma cholesterol levels [2]. FH is commonly caused by mutations in the low-density lipoprotein receptor (
Clinically, the key characteristics of patients with FH are severely elevated low-density lipoprotein cholesterol (LDL-c), premature corneal arcus (<45 years old), and tendon xanthomas, predominantly in the Achilles tendon and on the extensor tendons of the elbows, hands, knees, and toes [7,8]. FH can be identified and diagnosed using established clinical diagnostic criteria, such as Simon Broome (SB) criteria [9] and Dutch Lipid Clinic Criteria (DLCC) [10]. Genetic testing to confirm the diagnosis can be offered if resources are available [11].
HoFH is rarely reported [12], with a global prevalence estimated at 1 in 160 000 to 1 in 300 000 [7]. HeFH is more common, with a global prevalence estimated at 1 in 200 to 1 in 500 [13]. A recent systematic review and meta-analysis showed that the pooled prevalence of HeFH based on genetic testing or established clinical diagnostic criteria among the general adult population was 1 in 303 [14]. In Malaysia, the prevalence of clinically diagnosed HeFH was estimated to be 1 in 100 [15], while the prevalence of genetically confirmed HeFH was recently reported as 1 in 427 [16]. This is comparable to the prevalence in other countries [14].
If FH is left untreated, men with FH have a 50% risk of developing coronary artery disease (CAD) by the age of 50 years, and women with FH have a 30% risk of developing CAD by the age of 60 [17]. FH remains underdiagnosed and undertreated, especially in the primary care setting [13]. This results in a lost opportunity for preventing premature CAD [13,15]. Therefore, early identification in primary care of patients who are at risk of FH is pivotal. However, the established clinical criteria, such as SB and DLCC, have limited utility for the purposes of case-finding in primary care [18], as they were developed from secondary care registers of FH patients. These diagnostic criteria would require a comprehensive physical examination (tendon xanthoma and arcus cornealis), family history recording, and genetic mutation testing, which are not routinely assessed or are inadequately documented in primary care. As a consequence, researchers in the United Kingdom have developed a new case-finding tool, the Familial Hypercholesterolemia Case Ascertainment Tool (FAMCAT), based on a risk prediction algorithm developed and validated from primary care databases [18,19]. In Malaysia, genetic testing is not routinely available due to limited financial resources as well as a lack of expertise to conduct genetic testing. Therefore, the SB, DLCC, and FAMCAT can be used to identify FH cases in primary care when genetic testing is unavailable.
With or without genetic testing, patients with clinically diagnosed FH are at a very high risk to develop atherosclerotic cardiovascular diseases (ASCVD) and therefore should be treated with high-dose potent statins and therapeutic lifestyle modifications [20,21]. If patients fail to achieve an LDL-c target of <1.8 mmol/L, the current Malaysian clinical practice guideline recommends the combination of high-dose potent statins with either ezetimibe or
Case Reports
METHODOLOGY:
The 3 patients reported in this case series were participants of a study titled “Reducing Premature Coronary Artery Disease by Early Identification of Familial Hypercholesterolemia: The UK-Malaysia Joint-Partnership Call on Non-Communicable Diseases” (grant reference: 100-TNCPI/GOV 16/6/2 [002/2020]-02 and MR/T 017384/1).
These 3 patients were identified in the primary care setting from September 2020 to April 2021 and were invited to participate. A patient information sheet about the study was given, and written informed consent was obtained upon recruitment into the study. Written informed consent to use the clinical data and digital images was also obtained from all patients in this case series.
Demographic data (age, ethnicity, education level, marital status, household income, and family history) were obtained from the patients. Information on symptoms of angina and peripheral vascular disease (PVD) were collected using the WHO Rose Angina Questionnaire [24] and Edinburgh Claudication Questionnaire [25], respectively. Information on potential secondary causes of hypercholesterolemia, such as chronic kidney disease (CKD), diabetes, and hypothyroidism, were obtained from the patients and verified with their electronic medical records (EMR). Data on current medications and the highest ever recorded fasting serum lipid profiles were obtained from the EMR. Family history variables (elevated cholesterol level, FH, and myocardial infarction) were collected using a validated Family History Questionnaire, which has been translated into the Malay language. Physical examinations to identify tendon xanthomas and corneal arcus were conducted during the consultation.
Using these variables, the SB criteria, DLCC score, and FAMCAT relative risk score was assessed for each patient. They would be clinically diagnosed to have FH if they fulfilled “definite” or “possible” FH by SB criteria [9], DLCC score ≥6 (“definite” or “probable” FH) [10], or FAMCAT algorithm relative risk score >1 (high probability of FH) [26]. The 3 patients were found to fulfill at least 1 of the FH clinical diagnostic criteria. Therefore, they were offered genetic testing. Primary care physicians were trained by the lipid specialists to deliver genetic counseling to the patients prior to genetic testing. Patients were counseled regarding their risk of having FH, the pattern of inheritance, the genetic testing procedure, confirmation of diagnosis after the genetic testing, further management, and the need for cascade screening of first-degree relatives once they are genetically diagnosed. Written informed consent was obtained from the patients.
Approximately 10 mL of venous blood sample was collected from the patients into an ethylene diamine tetra-acetic acid tube at the primary care clinics. These samples were kept in a temperature-controlled container and delivered to the Universiti Teknologi MARA genetic laboratory within 3 h of collection. Once received, genomic DNA was extracted from the whole blood collected using a MasterPure DNA Purification Kit for Blood Version II (Epicentre, Madison, Winsconsin), according to the manufacturer’s protocol. The concentration and purity of extracted DNA were determined using a SpectraMax QuickDrop micro-volume spectrophotometer (Molecular Devices, San Jose, CA, USA) followed by a Qubit fluorometer (Thermo Fisher Scientific, Wilmington, DE, USA). The samples were then subjected to targeted next generation sequencing, covering all the exons (including the exon-intron boundaries) and the 5’ and 3’ untranslated regions of the 4 FH major genes (LDLR, APOB, PCSK9, and LDLRAP1). Library preparation was performed using an AmpliSeq Library PLUS kit (Illumina, San Diego, CA, USA), and enriched samples were sequenced on an ISeq 100 sequencer platform (Illumina, San Diego, CA, USA), according to the manufacturer’s instructions. A GRCh37 hg19 human reference assembly was used to map genomic sequencing data using the proprietary BaseSpace Sequence Hub application (Illumina, San Diego, CA, USA). A Germline Variant Caller (Illumina, San Diego, CA, USA) was used to perform variant calling. Only variants with minor allele frequency ≤5% in the 1000 Genomes database were assessed, using in silico web-based software, ClinVar and Leiden Open Variation Database, previous published reports, and clinical data from the 3 cases. The pathogenicity of the variants was agreed based on the American College of Medical Genetics and Genomics guidelines [27], by which variants were classified as pathogenic, likely pathogenic, benign, likely benign, or variants of uncertain significance. Likely pathogenic and pathogenic variants were collectively referred to as pathogenic variants.
Lipid specialists trained the primary care physicians to deliver genetic analysis results to the patients. During consultation, patients who were genetically confirmed to have FH were counseled regarding the nature of the genetic mutations, mode of inheritance, and need for further management, including cascade screening of first-degree relatives. The importance of adherence to lifestyle modification and pharmacotherapy were emphasized. Lipid-lowering treatment for each patient was reviewed and up-titrated to achieve an LDL-c target of <1.8 mmol/L [20,22]. Patients were also supported psychosocially on how to adapt with the condition. They were referred to the Universiti Teknologi MARA lipid specialists for further management and for the cascade screening of first-degree relatives.
CASE 1:
This patient was a 39-year-old Malay woman, working as a civil servant, who presented to a primary care clinic for routine follow-up for hypercholesterolemia. She was on atorvastatin 20 mg at night and therapeutic lifestyle modification, which included advice on dietary intake and physical activity. This patient was asymptomatic, with no personal history of hypertension, diabetes, or hypothyroidism. The WHO Rose Angina Questionnaire [24] and the Edinburgh Claudication Questionnaire [25] were negative. There was also no history of CAD, cerebrovascular disease (CVD), PVD, or CKD. She was a non-smoker and did not drink alcohol.
It was noted that she had a strong family history of elevated cholesterol level and premature CAD, particularly on the paternal side. Her father was suspected to have FH at the age of 42 years old. He had an episode of myocardial infarction (MI) at the age of 46 years old and eventually underwent coronary artery bypass grafting (CABG) 1 year after the MI, at the age of 47 years. He was currently 65 years old and was being treated with a
On the maternal side, the family history was uneventful, as her maternal grandfather had a minor stroke at the age of 82 years old. Despite these findings, none of her family members had undergone genetic testing. Her family pedigree chart is shown in Figure 1.
On examination, her vital signs were normal. There were tendon xanthomas on her bilateral metacarpophalangeal joints, Achilles tendon, and knees. There was no premature corneal arcus. Other physical examinations, including of the cardiovascular and respiratory systems, were unremarkable. Figure 2 shows the physical examination findings.
According to the data extracted from her EMR, the highest cholesterol level was noted in 2019, with an LDL-c of 8.6 mmol/L and a total cholesterol (TC) of 10.4 mmol/L. In terms of clinical diagnosis, this patient fulfilled the SB criteria (definite FH), DLCC (score of 16: definite FH), and FAMCAT (relative risk score of 32.3). Upon targeted next generation sequencing analysis, the patient was found to carry a frameshift c.660del pathogenic variant in the
CASE 2:
This patient was an unemployed 57-year-old Chinese woman, diagnosed with hypercholesterolemia at the age of 55 years, and with no other known medical illness. She presented at a primary care clinic for a routine follow-up. This patient was on simvastatin 20 mg at night and received advice on dietary in-take and physical activity. During this visit, she was assessed using the WHO Rose Angina Questionnaire [24] and described having a history of central chest tightness at the age of 55 years, which resolved in less than 5 min. The pain was worse after a meal and was not exacerbated by exertion or physical activity. She never had any chest pain at rest or any chest discomfort that lasted more than 30 min. She did not seek medical attention at the time, and it was not further investigated. She was also assessed using the Edinburgh Claudication Questionnaire [25] and was found to be negative for the presence of PVD symptoms. There was no personal history of secondary causes of hypercholesterolemia, such as diabetes, hypertension, or hypothyroidism. There was no history of CAD, CVD, PVD, or CKD recorded in her EMR. She drank alcohol occasionally but did not smoke cigarettes.
This patient has a significant family history of hypercholesterolemia. Her father, elder sister, younger brother, and younger sister were all on lipid-lowering therapy. She noted that her elder sister had xanthelasma around the eyes and premature corneal arcus in her forties. Her younger brother also had premature corneal arcus in his forties. Her mother died of a recurrent stroke at the age of 56 years old; the first stroke occurred at the age of 54 years. Figure 3 depicts the patient’s family pedigree chart.
On examination, she was well and did not appear to be in pain. Her vital signs were normal. Stage 4 corneal arcus was present in both eyes, as shown in Figure 4; she noticed that they started in her thirties. Other physical examinations were unremarkable.
According to the data extracted from her EMR, the highest cholesterol level was noted in 2019, with an LDL-c of 5.7 mmol/L and TC of 8.5 mmol/L. Based on these findings, this patient fulfilled the SB criteria (possible FH), DLCC (score of 14: definite FH), and FAMCAT (relative risk score of 4.4). The genetic analysis of this patient showed a missense c.10579C>T pathogenic variant in the
This patient was immediately referred to the family medicine specialist at the primary care clinic for further assessment of her chest pain. Her resting electrocardiogram was normal, and she was given a follow-up appointment with the family medicine specialist for further monitoring of her chest pain. Despite pharmacological therapy and therapeutic lifestyle modification, her LDL-c and TC levels remained high and she failed to achieve the LDL-c target of <1.8 mmol/L. Therefore, she was also referred to the lipid specialists for further management. Cascade screening of first-degree relatives is currently being conducted by the lipid specialists in order to identify those affected with FH.
CASE 3:
This patient was a 41-year-old Malay man with underlying hypertension. He was working as a government official. He came to a primary care clinic for a routine follow-up. He was asymptomatic, with no subclinical evidence of ASCVD. There was no personal history of hypercholesterolemia, diabetes, or hypothyroidism. The WHO Rose Angina Questionnaire [24] and the Edinburgh Claudication Questionnaire [25] were negative. There was no history of CAD, CVD, PVD, or CKD recorded in the EMR. This patient had stopped smoking in 2016. He had smoked 10 pack years of cigarettes since he was 18 years old. His mother had a stroke at 60 years of age. There were no other family members with history of hypercholesterolemia, CAD, CVD, or PVD. His family pedigree chart is shown in Figure 5.
On examination, his vital signs were normal. There was no tendon xanthoma or premature corneal arcus. Other physical examinations, including the cardiovascular and respiratory systems, were unremarkable.
According to the data extracted from his EMR, the highest cholesterol level was noted in 2020, with an LDL-c of 5.4 mmol/L and a TC of 7.3 mmol/L. This patient fulfilled the SB criteria (possible FH) but did not fulfill the DLCC (score of 4) and FAMCAT (relative risk score of 0.6). The genetic analysis of this patient showed a missense c.277C>T mutation in the PCSK9 gene (rs151193009), which was located within exon 2. There are conflicting findings whether this particular mutation is pathogenic, a variant of uncertain significance, or benign [28–31].
He was initially managed in primary care with simvastatin 10 mg at night and therapeutic lifestyle modification, which included advice on dietary intake and physical activity. However, he failed to achieve the LDL-c target of <1.8 mmol/L and was therefore referred to the lipid specialists for further management. Cascade screening of first-degree relatives is currently being conducted by the lipid specialists.
Table 1 shows a summary of all 3 patients selected for this case series, who were identified in primary care and had subsequently been genetically tested.
Discussion
This case series is the first to report genetically confirmed HeFH in the Malaysian primary care setting. We presented 3 cases of patients aged 39 to 57 years old with
In Asian populations, including Malaysian, the most common FH mutation that has been described is of the
With regards to LDL-c levels, the patients presented in this case series had high levels of LDL-c, ranging from 5.4 to 8.6 mmol/L, which is 3 to 5 times higher than the target value of 1.8 mmol/L for individuals at very high cardiovascular risk [22]. This finding is similar to that of a Canadian study involving 313 genetically confirmed HeFH patients, in which 42% of the patients had LDL-c levels between 5.0 and 6.0 mmol/L, and 88% of the patients had an LDL-c level above 8.0 mmol/L [37]. It was observed that the patient in case 1, who was confirmed to have an
With regards to the p.Arg3527Trp pathogenic variant that is located in the
The
A higher LDL-c level in the blood has been associated with a more severe expression of phenotypes [46]. Phenotypically, we found that the patient in case 1, who was confirmed to have
FH is still currently underdiagnosed in Malaysia, with a detection rate estimated at 0.5% [34]. Primary care physicians should have a high index of suspicion in patients presenting with LDL-c levels of >4.9 mmol/L [22,23]. In this case series, primary care physicians were trained on how to perform a thorough physical examination and to be familiar with the appropriate clinical diagnostic tools, such as the SB, DLCC, and FAMCAT case-finding tool. Referral for genetic testing should be considered if the patient fulfills the clinical diagnostic criteria and if genetic testing is routinely available, as this is the criterion standard to diagnose FH [48]. As with many developing countries, genetic testing is not routinely available in Malaysia owing to limited financial resources. However, in this case series, patients who fulfilled at least 1 of the FH clinical diagnostic criteria were offered genetic testing, which was funded by the study. Primary care physicians were trained by the lipid specialists to deliver genetic counseling to the patients prior to genetic testing. The counseling included informing the patients of their risk of having FH, the pattern of inheritance, genetic testing procedure, confirmation of diagnosis after the genetic testing, further management, and need for cascade screening of first-degree relatives once they are genetically diagnosed.
Primary care physicians were also trained to deliver genetic analysis results to the patients. In this cases series, once the patients were confirmed to have FH, they were counseled regarding the nature of the genetic mutations and mode of inheritance. These patients’ cases were initially managed in primary care with statins and therapeutic lifestyle modification, which included advice on dietary intake and physical activity. Patients with FH are recommended to receive individualized advice about diet, physical activity, and healthy weight maintenance from healthcare providers to reduce their cardiovascular risks [49–51]. With regards to the pharmacological management for patients with FH, the current Malaysian clinical practice guideline recommends the combination of high-dose potent statins with either ezetimibe or
Apart from pharmacological treatment with lipid-lowering medications, cascade screening of first-degree relatives should also be conducted to allow early detection and management of this autosomal dominant disorder. Cascade screening is a cost-effective method to identify additional cases of FH through systematic family tracing [54]. In this case series, primary care physicians also played a significant role in identifying first-degree relatives to be referred for cascade testing. Early detection of FH will reduce the average age at which family members are diagnosed with FH and increase the percentage of them receiving pharmacological treatment [54]. In addition to the above measures, long-term follow-up by both a primary care physician and lipid specialist is vital to ensure patients with FH achieve the LDL-c target, hence preventing premature ASCVD. The need to integrate the pivotal role of primary care physicians with that of the lipid specialists’ service to enhance the detection and management of FH in the community is well recognized [55]. There is extensive evidence to show that this approach is clinically and financially effective [56–58].
Additionally, accurate national registries on the prevalence of FH would assist in increasing the awareness of this condition among clinicians [59]. In Malaysia, a national level FH registry is currently being established [60]. Clinical practice guidelines on the management of FH in Malaysia should also be established to improve early detection and treatment of FH, especially in primary care, which serves as the front-line of the healthcare setting. It is hoped that these systematic and widespread efforts will improve the prevention of premature ASCVD in this population.
Conclusions
In summary, we reported 3 cases of patients with mutations of
References:
1.. Pejic RN, Familial hypercholesterolemia: Ochsner J, 2014; 14; 669-72
2.. Vrablik M, Tichý L, Freiberger T, Genetics of familial hypercholesterolemia: New insights: Front Genet, 2020; 11; 574474
3.. Doi T, Hori M, Harada-Shiba M, Patients with LDLR and PCSK9 gene variants experienced higher incidence of cardiovascular outcomes in heterozygous familial hypercholesterolemia: J Am Heart Assoc, 2021; 10; e018263
4.. Truong PK, Lao TD, Le THA, The major molecular causes of familial hypercholesterolemia: Asian J Pharma Res Health Care, 2018; 10; 60-68
5.. Garcia CK, Wilund K, Arca M, Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein: Science, 2001; 292; 1394-98
6.. Asselbergs FW, Guo Y, van Iperen EP, Large-scale gene-centric meta-analysis across 32 studies identifies multiple lipid loci: Am J Hum Genet, 2012; 91; 823-38
7.. Alnouri F, Al-Allaf FA, Athar M, Xanthomas can be misdiagnosed and mistreated in homozygous familial hypercholesterolemia patients: A call for increased awareness among dermatologists and health care practitioners: Glob Heart, 2020; 15; 19
8.. Bouhairie VE, Goldberg AC, Familial hypercholesterolemia: Cardiol Clin, 2015; 33; 169-79
9.. , Mortality in treated heterozygous familial hypercholesterolaemia: Implications for clinical management [Scientific Steering Committee on behalf of the Simon Broome Register Group]: Atherosclerosis, 1999; 142; 105-12
10.. Watts GF, Sullivan DR, Poplawski N, Familial hypercholesterolaemia: A model of care for Australasia: Atheroscler Suppl, 2011; 12; 221-63
11.. Qureshi N, Akyea RK, Dutton B, Case-finding and genetic testing for familial hypercholesterolaemia in primary care: Heart, 2021; 107; 1956-61
12.. Alicezah MK, Razali R, Rahman T, Homozygous familial hypercholesterolemia: Malays J Pathol, 2014; 36; 131-17
13.. Nordestgaard BG, Chapman MJ, Humphries SE, Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society [published erratum appears in Eur Heart J. 2020;41:4517]: Eur Heart J, 2013; 34; 3478-90a
14.. Hu P, Dharmayat KI, Stevens CAT, Prevalence of familial hypercholesterolemia among the general population and patients with atherosclerotic cardiovascular disease: A systematic review and meta-analysis: Circulation, 2020; 22; 1742-59
15.. Vallejo-Vaz AJ, De Marco M, Stevens CAT, Overview of the current status of familial hypercholesterolaemia care in over 60 countries – The EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC): Atherosclerosis, 2018; 277; 234-55
16.. Razman AZ, Chua YA, Mohd Kasim NA, Genetic spectrum of familial hypercholesterolaemia in the malaysian community: Identification of pathogenic gene variants using targeted next-generation sequencing: Int J Mol Sci, 2022; 23; 14971
17.. Goldberg AC, Hopkins PN, Toth , Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: Clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia: J Clin Lipidol, 2011; 5(3 Suppl.); S1-8
18.. Qureshi N, Weng S, Tranter J, Feasibility of improving identification of familial hypercholesterolaemia in general practice: Intervention development study [published erratum appears in BMJ Open. 2016;: e011734corr1]: BMJ Open, 2016; 5; e011734
19.. Weng SF, Kai J, Neil HA, Improving identification of familial hyper-cholesterolaemia in primary care: Derivation and validation of the familial hypercholesterolaemia case ascertainment tool (FAMCAT): Atherosclerosis, 2015; 2; 336-43
20.. : Identification and Management of Familial Hypercholesterolemia, 2019, United Kingdom: NICE Clinical Guideline
21.. Gidding SS, Champagne MA, de Ferranti SD, A scientific statement from the American Heart Association: Circulation, 2015; 22; 2167-92
22.. : 5th Edition of Clinical Practice Guidelines: Management of Dyslipidaemia, 2017; 20-21
23.. Kasim NAM, Chua YA, An updated overall management: J Clin Health Sci, 2020; 5; 19-38
24.. Rahman MA, Spurrier N, Mahmood MA, Rose Angina Questionnaire: Validation with cardiologists’ diagnoses to detect coronary heart disease in Bangladesh: Indian Heart J, 2013; 65; 30-39
25.. Boylan L, Nesbitt C, Wilson L, Reliability of the Edinburgh Claudication Questionnaire for identifying symptomatic PAD in general practice: Angiology, 2021; 72; 474-79
26.. Weng S, Kai J, Akyea R, Qureshi N, Detection of familial hypercholesterolaemia: external validation of the FAMCAT clinical case-finding algorithm to identify patients in primary care [published erratum appears in Lancet Public Health. 2019;4(7)e325]: Lancet Public Health, 2019; 4; e256-e64
27.. Richards S, Aziz N, Bale S, Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology: Genet Med, 2015; 17; 405-24
28.. Lee C, Cui Y, Song J, Effects of familial hypercholesterolemia-associated genes on the phenotype of premature myocardial infarction: Lipids Health Dis, 2019; 18; 95
29.. Yang L, Pu T, Zhang Y, The R93C variant of PCSK9 reduces the risk of premature MI in a Chinese Han population: Front Genet, 2022; 13; 875269
30.. Hsu LA, Teng MS, Wu S, Common and rare PCSK9 variants associated with low-density lipoprotein cholesterol levels and the risk of diabetes mellitus: A Mendelian randomization study: Int J Mol Sc, 2022; 18; 10418
31.. Yu H, Pu T, Xu M, Gao W, P808 Association between genetic variants in PCSK9/APOB/LDLR and premature myocardial infarction in Han Chinese: Eur Heart J, 2018; 39(Suppl. 1); ehy564-P808
32.. Al-Khateeb A, Al-Talib H, Genetic researches among Malaysian familial hypercholesterolaemic population: JUMMEC, 2016; 19; 1-11
33.. Jackson CL, Zordok M, Kullo IJ, Familial hypercholesterolemia in Southeast and East Asia: Am J Prev Cardiol, 2021; 6; 100157
34.. Chua YA, Razman AZ, Ramli AS, Familial hypercholesterolaemia in the Malaysian community: Prevalence, under-detection and under-treatment: J Atheroscler Thromb, 2021; 28; 1095-107
35.. Huang CC, Charng MJ, Genetic diagnosis of familial hypercholesterolemia in Asia: Front Genet, 2020; 11; 833
36.. Hori M, Ohta N, Takahashi A, Impact of LDLR and PCSK9 pathogenic variants in Japanese heterozygous familial hypercholesterolemia patients: Atherosclerosis, 2019; 289; 101-8
37.. Wang J, Dron JS, Ban MR, Polygenic versus monogenic causes of hypercholesterolemia ascertained clinically: Arterioscler Thromb Vasc Biol, 2016; 36; 2439-45
38.. Fahed AC, Nemer GM, Familial hypercholesterolemia: The lipids or the genes?: Nutr Metab (Lond), 2011; 8; 23
39.. Mabuchi H, Nohara A, Noguchi T, Genotypic and phenotypic features in homozygous familial hypercholesterolemia caused by proprotein convertase subtilisin/kexin type 9 (PCSK9) gain-of-function mutation: Atherosclerosis, 2014; 236; 54-61
40.. Goldstein JL, Brown MS, Regulation of low-density lipoprotein receptors: Implications for pathogenesis and therapy of hypercholesterolemia and atherosclerosis: Circulation, 1987; 76; 504-7
41.. Bamimore MA, Zaid A, Banerjee Y, Familial hypercholesterolemia mutations in the Middle Eastern and North African region: A need for a national registry: J Clin Lipidol, 2015; 9; 187-94
42.. Hu M, Hooper AJ, Bockxmeer FM, Management of familial hypercholesterolemia in Hong Kong: J Atheroscler Thromb, 2016; 23; 520-31
43.. : ClinVar [VCV000226330.13]. [cited 2022 Dec 28]. Available from: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000226330.13
44.. Al-Khateeb A, Al-Talib H, Mohamed MS, Phenotype-genotype analyses of clinically diagnosed Malaysian familial hypercholestrolemic patients: Adv Clin Exp Med, 2013; 22; 57-67
45.. Ejarque I, Real JT, Martinez-Hervas S, Evaluation of clinical diagnosis criteria of familial ligand defective apoB 100 and lipoprotein phenotype comparison between LDL receptor gene mutations affecting ligand-binding domain and the R3500Q mutation of the apoB gene in patients from a South European population: Transl Res, 2008; 151; 162-67
46.. Al-Khateeb AR, Mohd MS, Yusof Z, Zilfalil BA, Molecular description of familial defective APOB-100 in Malaysia: Biochem Genet, 2013; 51; 811-23
47.. Nishikawa T, Tada H, Nakagawa-Kamiya T, A case with familial hypercholesterolemia complicated with severe systemic atherosclerosis intensively treated for more than 30 years: J Cardiol Cases, 2020; 22; 216-20
48.. Futema M, Taylor-Beadling A, Williams M, Humphries SE, Genetic testing for familial hypercholesterolemia-past, present, and future: J Lipid Res, 2021; 62; 100139
49.. Kinnear FJ, Hamilton-Shield JP, Stensel DJ, Nutrition and physical activity intervention for families with familial hypercholesterolaemia: Protocol for a pilot randomised controlled feasibility study: Pilot Feasibility Stud, 2020; 6; 42
50.. Watts GF, Gidding S, Wierzbicki AS, Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation: Int J Cardiol, 2014; 171; 309-25
51.. Catapano AL, Graham I, De Backer G, 2016 ESC/EAS Guidelines for the management of dyslipidaemias: Eur Heart J, 2016; 37; 2999-3058
52.. Azraii AB, Ramli AS, Ismail Z, Knowledge, awareness and practice regarding familial hypercholesterolaemia among primary care physicians in Malaysia: The importance of professional training: Atherosclerosis, 2018; 277; 508-16
53.. Batais MA, Almigbal TH, Bin Abdulhak AA, Assessment of physicians’ awareness and knowledge of familial hypercholesterolemia in Saudi Arabia: Is there a gap?: PLoS One, 2017; 12; e0183494
54.. Knowles JW, Rader DJ, Khoury MJ, Cascade screening for familial hypercholesterolemia and the use of genetic testing: JAMA, 2017; 318; 381-82
55.. Arnold-Reed DE, Brett T, Troeung L, Detection and management of familial hypercholesterolaemia in primary care in Australia: Protocol for a pragmatic cluster intervention study with pre-post intervention comparisons: BMJ Open, 2017; 10; e017539
56.. Watts GF, Juniper A, van Bockxmeer F, Familial hypercholesterolaemia: A review with emphasis on evidence for treatment, new models of care and health economic evaluations: Int J Evid Based Healthc, 2012; 10; 211-21
57.. Ademi Z, Watts GF, Pang J, Cascade screening based on genetic testing is cost-effective: Evidence for the implementation of models of care for familial hypercholesterolemia: J Clin Lipidol, 2014; 8; 390-400
58.. Nherera L, Marks D, Minhas R, Probabilistic cost-effectiveness analysis of cascade screening for familial hypercholesterolaemia using alternative diagnostic and identification strategies: Heart, 2011; 97; 1175-81
59.. Amerizadeh A, Javanmard SH, Sarrafzadegan N, Vaseghi G, Familial hyper-cholesterolemia (FH) registry worldwide: A systematic review: Curr Probl Cardiol, 2022; 47; 100999
60.. Firus Khan AY, Ramli AS, Abdul Razak S, The Malaysian HEalth and WellBeing AssessmenT(MyHEBAT) study protocol: An initiation of a national registry for extended cardiovascular risk evaluation in the community: Int J Environ Res Public Health, 2022; 19; 11789
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133