24 April 2024: Articles 
Pathogenic Potential of a PCK1 Gene Variant in Cytosolic PEPCK Deficiency: A Compelling Case Study
Challenging differential diagnosis, Management of emergency care, Rare disease, Congenital defects / diseases
Monika Duś-ŻuchowskaDOI: 10.12659/AJCR.943118
Am J Case Rep 2024; 25:e943118






Abstract
BACKGROUND: Cytosolic phosphoenolpyruvate carboxykinase (PEPCK-C) deficiency is an extremely rare autosomal recessive inherited error of metabolism in which gluconeogenesis is impaired, resulting in life-threatening episodes of hypoglycemia and metabolic acidosis. The diagnosis of gluconeogenesis disorders is challenging. In the diagnostic pathway, the molecular test plays a paramount role.
CASE REPORT: The aim of the paper is to present the case report of a girl with recurrent episodes of severe hypoglycemia, in whom molecular diagnosis enabled the confirmation of PEPCK – C deficiency. The patient experienced 4 episodes of severe hypoglycemia. Most of them were accompanied by hyperlacticaemia, metabolic acidosis, and elevated liver enzymes. All of the metabolic decompensations were triggered by infectious agents. The episodes resolved after continuous infusion of high-dose glucose. Due to the recurrent character of the disease, a genetic condition was suspected. The differential diagnosis included metabolic and endocrinological causes of hypoglycemia. Two variants in the PCK1 gene were detected: c.265G>A p.(Glu89Lys) in exon 3 and c.925G>A p.(Gly309Arg) in exon 6. As c.925G>A p.(Gly309Arg) is a known pathogenic variant, the second variant was first described in June 2023 in the ClinVar database and described as “with unknown clinical significance”.
CONCLUSIONS: According to the clinical symptoms observed in the presented case, the variant c.265G>A p.(Glu89Lys) in PCK1 gene should be considered likely pathogenic. We suggest considering molecular diagnostics in every patient presented with recurrent, severe hypoglycemia with accompanying liver damage as most accurate, feasible, and reliable method.
Keywords: Heterozygote, Hypoglycemia, Phosphoenolpyruvate carboxykinase deficiency, Female, Humans, Gluconeogenesis, Intracellular Signaling Peptides and Proteins, Phosphoenolpyruvate Carboxykinase (GTP)
Introduction
Cytosolic phosphoenolpyruvate carboxykinase (PEPCK-C) deficiency is an extremely rare defect of gluconeogenesis, which is the process needed to produce energy during fasting.
In humans, substrates for gluconeogenesis come from non-carbohydrate precursors of pyruvate or intermediates of citric acid. PEPCK-C is involved in the process of gluconeogenesis by catalyzing the conversion of oxaloacetate into phosphoenolpyruvate, which is then transformed to glucose via a series of enzymes.
The main signs of this disease are episodes of hypoglycemia and lactic acidosis. Hypo- or hyperketosis, high urine excretion of Krebs cycle metabolites (particularly fumarate), and hepatic function disturbances have also been described. The second isoform of phosphoenolpyruvate carboxykinase, mitochondrial (PEPCK-M), is not affected [1,2].
In humans, PEPCK-C is encoded by
Disorders of gluconeogenesis enzymes are very rare. Their overall prevalence is less than 1: 50 000 [6]. The incidence of PEPCK deficiency is unknown [3]. The leading sign of gluconeogenesis defects is hypoglycemia. As hypoglycemia is a common symptom of many inherited errors of metabolism, the exact diagnosis requires a broad spectrum of laboratory tests for endocrinological and metabolic causes, among which gluconeogenesis disorders are very rare but treatable conditions. A precise diagnosis of gluconeogenesis disorders can be established using molecular analysis [7].
Goetz et a. reported on 32 patients from 25 families with a confirmed genetic diagnosis of PEPCK-C deficiency. The most common variant detected in the
In this article, we present the case of symptomatic PEPCK-C deficiency in a girl who was compound heterozygote with a known common variant and the second variant described recently in June 2023 as “with unknown clinical significance” in the
Case Report
A 12-month-old girl was transported to the hospital by a medical rescue team due to disturbed consciousness. She was from the first pregnancy, born by natural birth, at term, with a birth weight of 3630 g and an Apgar score of 10. She was vaccinated according to the vaccination schedule. Her psycho-motor development was normal. One day before admission to the hospital, she had a body temperature of 37.8°C, and for approximately 16 h she refused to eat or drink. The mother called an ambulance as the daughter presented nystagmus and was unresponsive.
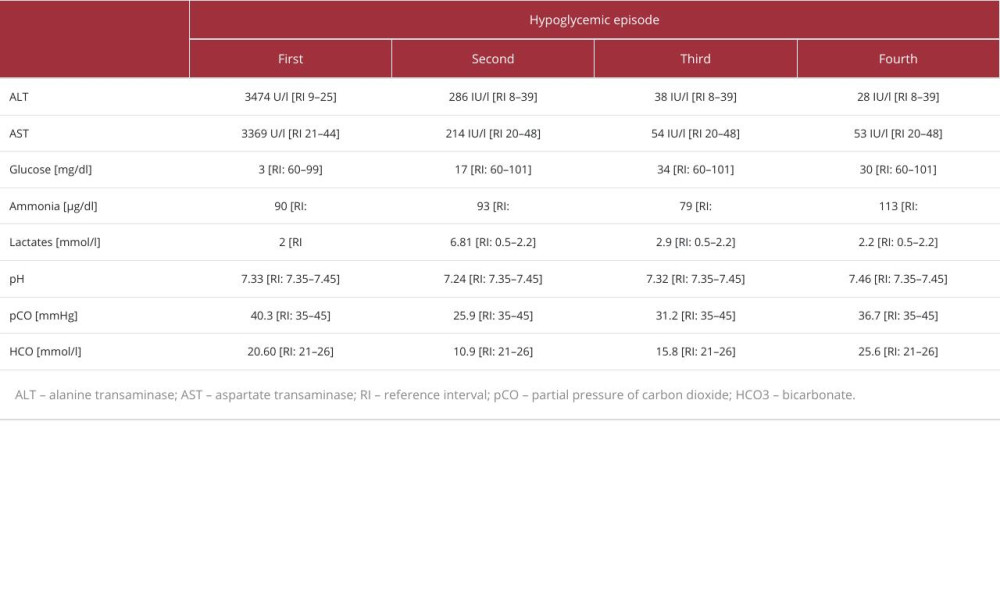
At admission to the hospital, her Glasgow Coma Scale score was 11. She had positive meningeal symptoms. The laboratory tests revealed severe hypoglycemia (3 mg/dl), metabolic acidosis with slightly elevated levels of lactate, and highly abnormal liver function test results (Table 1). The urine test for ketones was negative. Laboratory diagnostics for infectious causes such as human immunodeficiency virus (HIV), hepatitis B Virus (HBV), cytomegalovirus (CMV),
Antibacterial treatment and 10% glucose continuous intravenous infusion in the dose of 50 ml/h (0.5 g/kg body weight/hour) were administered to prevent metabolic decompensation. As the girl required continuous infusion of glucose to prevent recurrent episodes of hypoglycemia, the endocrino-logic and metabolic causes were taken into consideration. The cortisol concentration (62.0; 77.0 ng/ml) corresponding to the reference norm for age excluded adrenal insufficiency as a potential cause of hypoglycemia.
Although slightly elevated insulin (20.6 uU/mL) levels were initially noted during continuous glucose infusion, taking into account the lack of ketones and severe episodes of hypoglycemia recurring when glucose infusion was discontinued, it was decided to perform diagnostics for hyperinsulinism. After the child’s metabolic control, an 8-h fasting test was performed – glycemia remained within normal limits (78 mg/dl; 66 mg/dl), with low insulin output (0.9 uU/ml; <1 uU/ml).
The diagnostic process was extended to metabolic diseases. In the profile of the organic acids in urine (GC/MS), ketonuria with dicarboxylic aciduria (glutaric and adipic acid), increased 3-hydroxydicarboxylic acid C6–C12 aciduria, and increased excretion of fumaric and malic acid were detected. The profile of acylcarnitines by tandem mass spectrometry (MS/MS) revealed non-specific elevations of C2–C18 acylcarnitines.
In the differential diagnosis, the glycogen storage diseases (GSDs) were suspected; therefore, a sweet meal and an intravenous glucose load test (0.5 g/kg body weight) were performed administered, but the results did not reveal specific changes in glucose, lactate concentrations, or in blood-gas analysis characteristic for classical hepatic GSDs.
Approximately 6 months later, during the infection, the girl presented a second episode of hypoglycemia (17 mg/dl) with seizures. Similar to the previous episode of hypoglycemia, she had metabolic acidosis and high levels of lactate, alanine transaminase (ALT), and aspartate transaminase (AST). Additionally, a slightly elevated level of ammonia and an increased concentration of free fatty acids were observed (Table 1). Treatment with glucose infusion and antibiotic therapy was applied. The girl required continuous glucose infusion (0.35–0.4 g/kg body weight/hour) to maintain glucose level >100 mg/ml.
Due to episodes of recurrent decompensations with severe hypoglycemia of unclear origin, the girl was referred to our Metabolic Diseases Department. There, a prolonged fasting test was performed, during which the levels of lactate increased (23.8 [RI: 4.5–19.8] mg/dl). The concentration of hydroxybutyric acid was also determined, and the result was within the normal range (0.42 [RI0.03–0.65] mmol/l). Based on clinical symptoms, glycogen synthase deficiency was suspected. Blood was collected for NGS genetic tests. After a dietary consultation, to avoid prolonged fasting, an additional night feeding was introduced.
While waiting for the genetic test results, the girl had a third episode of symptomatic hypoglycemia at age 24 months, likely caused by an upper respiratory tract infection. Laboratory tests showed metabolic acidosis and high levels of lactate, AST, and C-reactive protein (CRP) (Table 1). Her condition improved after antibiotic therapy and glucose supplementation.
Wigh probability, the genetic test ruled out the presence of glycogen synthase deficiency. In the meantime, PEPCK deficiency was suspected. The targeted gene panel (TruSight One from Illumina) sequencing revealed 2 molecular variants in the
The c.265G>A p.(Glu89Lys) variant causes a missense change involving alteration of a conserved nucleotide (PhyloP100=9.46). The variant allele was found at a frequency of 0.0000263 in 152 208 control chromosomes in the gnomAD database, with no homozygous occurrence. It was described in June 2023 in the ClinVar database, but the clinical significance was unclear due to insufficient data. In-silico tools (Meta RNN, CADD, FATHMMKL, Mutation Assessor, SIFT, PolyPhen 2 HDIV and HVAR, Mutation Taster) predict a deleterious outcome for this variant. According to ACMG (American College of Medical Genetics and Genomics) criteria, this variant has been classified as likely pathogenic (PM3+PP3_moderate+PM2_supporting+PP4+BP1_ supporting=5 points). Moreover, the clinical symptoms observed in the presented case seem to confirm the potentially pathogenic nature of the aforementioned variant.
c.925G>A p.(Gly309Arg) is a common pathogenic variant that has been reported in the ClinVar and LOVD databases in correlation with PEPCK-C deficiency. In addition, there is functional evidence for the pathogenicity of this change. Western blot testing after overexpression in COS-1 cells showed less abundant mutant protein compared to wild-type PCK1, and PEPCK activity of the p.Gly309Arg mutant in transfected COS-1 cells was significantly lower than that of wild-type [2].
From 2016, cornstarch therapy, used at night in the prevention of hypoglycemia, was recommended. Since the initiation of this treatment, only 1 episode of hypoglycemia, in the course of respiratory tract infection, has been observed in the child. She remains under the constant care of specialists in metabolic pediatrics, her psychomotor development is normal, and she did not experience any further decompensations.
Discussion
Genetic confirmation of diagnosis is essential in diagnosis of PEPCK deficiency [3]. To date, 32 cases of genetically confirmed PEPCK-C deficiency with biallelic variants in
Our patient had PEPCK-C deficiency, with 2 detected variants in the
PEPCK-C deficiency can manifest itself in the neonatal period. The average age of patients who develop the disease after the neonatal period is 2.6 years [1]. In our patient, the first episode took place exactly on her first birthday.
The clinical picture of this disease can be mild, but an episode of hypoglycemia can also be very dangerous or even fatal [1,11]. Our patient had no symptoms of the disease apart from 4 episodes of symptomatic hypoglycemia, and her episodes of hypoglycemia were induced by infection or prolonged fasting. The other triggers of hypoglycemic episodes include alcohol consumption, heavy physical exertion, and other catabolic states such as lactation and breastfeeding of a newborn [12].
Our patient had high levels of ammonia during some episodes of hypoglycemia. Cases of high ammonia levels with PEPCK-C deficiency were also described by Santra et al [13] and Oishi et al [14]. In addition to the role of PEPCK-C in gluconeogenesis and glyceroneogenesis, it is also the major cataplerotic enzyme that helps remove citric acid cycle anions left after the entry of the carbon skeletons of amino acids in the Krebs cycle. Hakimi et al suggests that in PEPCK-C deficiency, some amino acids accumulate in the blood due to inability to enter the citric acid cycle and due to de-novo synthesis from citric acid cycle intermediates accumulated in the liver. Hakimi et al reported that an elevated urea cycle flux to dispose of amino nitrogen from the breakdown of amino acids resulted in the increased concentration of ammonia in the blood of PEPCK-C-/- mice [15].
During the first episode of hypoglycemia in our patient, the profile of organic acids determined in urine revealed ketonuria with dicarboxylic aciduria (glutaric and adipic acid), increased 3-hydroxydicarboxylic acid C6–C12 aciduria, and increased excretion of fumaric and malic acid. This finding is consistent with Vieira et al, who described the presence of dicarboxylic and 3-hydroxydicarboxylic acids and Krebs cycle metabolites in the urine of patients with PEPCK-C deficiency, particularly fumarate. The increase in the level of free fatty acids observed in our patient is also worth considering. In healthy subjects, PEPCK-C is involved in glyceroneogenesis. The enzyme enhances the production of glycerol-3-phosphate during fasting and enables further re-esterification of lipolytic fatty acids into triglycerides, which can be stored in adipose tissue. In PEPCK-C deficiency the glyceroneogenesis is disturbed, which can result in an increase of free fatty acids. This deviation of free fatty acids concentration was observed in the present case and by Veira et al [2,16].
Conclusions
In our patient, the c.265G>A p.(Glu89Lys) variant in the
References:
1.. Goetz M, Schröter J, Dattner T, Genotypic and phenotypic spectrum of cytosolic phosphoenolpyruvate carboxykinase deficiency: Mol Genet Metab, 2022; 137(1–2); 18-25
2.. Vieira P, Cameron J, Rahikkala E, Novel homozygous PCK1 mutation causing cytosolic phosphoenolpyruvate carboxykinase deficiency presenting as childhood hypoglycemia, an abnormal pattern of urine metabolites and liver dysfunction: Mol Genet Metab, 2017; 120(4); 337-41
3.. Leonard JV, Phosphoenolpyyruvate carboxykinase deficiency: Encyclopedia of molecular mechanisms of disease, 2009; 1638, Berlin; Heidelberg, Springer
4.. Hanson RW, Garber AJ, Phosphoenolpyruvate carboxykinase. I. Its role in gluconeogenesis: Am J Clin Nutr, 1972; 25(10); 1010-21
5.. Adams DR, Yuan H, Holyoak T, Three rare diseases in one Sib pair: RAI1, PCK1, GRIN2B mutations associated with Smith-Magenis syndrome, cytosolic PEPCK deficiency and NMDA receptor glutamate insensitivity: Mol Genet Metab, 2014; 113(3); 161-70
6.. Coffee EM, Tolan DR, Gluconeogenesis: Inborn errors of metabolism: From neonatal screening to metabolic pathways, 2015; 68-91, New York, Oxford University Press
7.. Weinstein DA, Steuerwald U, De Souza CFM, Derks TGJ, Inborn errors of metabolism with hypoglycemia: Glycogen storage diseases and inherited disorders of gluconeogenesis: Pediatr Clin North Am, 2018; 65(2); 247-65
8.. Vidnes J, Sovik O, Gluconeogenesis in infancy and childhood. III. Deficiency of the extramitochondrial form of hepatic phosphoenolpyruvate carboxykinase in a case of persistent neonatal hypoglycaemia: Acta Paediatr Scand, 1976; 65(3); 307-12
9.. Hommes FA, Bendien K, Elema JD, Two cases of phosphoenolpyruvate carboxykinase deficiency: Acta Paediatr Scand, 1976; 65(2); 233-40
10.. Fiser RH, Melsher HL, Fisher DA, Hepatic phoshoenol pyruvate carboxykinase (PEPCK) deficiency – a new cause of hypoglycaemia in childhood: Pediatr Res, 1974; 8(4); 432
11.. Becker J, Haas NA, Vlaho S, Cytosolic phosphoenolpyruvate carboxykinase deficiency: Cause of hypoglycemia-induced seizure and death: Neuropediatrics, 2021; 52(5); 398-402
12.. Vieira P, Nagy II, Rahikkala E, Cytosolic phosphoenolpyruvate carboxykinase deficiency: Expanding the clinical phenotype and novel laboratory findings: J Inherit Metab Dis, 2022; 45(2); 223-34
13.. Santra S, Cameron JM, Shyr C, Cytosolic phosphoenolpyruvate carboxykinase deficiency presenting with acute liver failure following gastroenteritis: Mol Genet Metab, 2016; 118(1); 21-27
14.. Oishi K, Siegel C, Cork EE, Novel missense variants in PCK1 gene cause cytosolic PEPCK deficiency with growth failure from inadequate caloric in-take: J Hum Genet, 2021; 66(3); 321-25
15.. Hakimi P, Hohnson MT, Yang J, Phosphoenolpyruvate carboxykinase and the critical role of cataplerosis in the control of hepatic metabolism: Nutr Metab (Lond), 2005; 21(2); 33
16.. Hanson RW, Thematic minireview series: A perspective on the biology of phosphoenolpyruvate carboxykinase 55 years after its discovery: J Biol Chem, 2009; 284(40); 27021-23
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report  22,759,844
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133