08 March 2021: Articles 
Diagnosis of GATA2 Deficiency in a Young Woman with Hemophagocytic Lymphohistiocytosis Triggered by Acute Systemic Cytomegalovirus Infection
Challenging differential diagnosis, Rare disease
Nicole Burak1EF*, Naveed Jan2EF, Jason Kessler3EF, Erwin Oei4EF, Priya Patel5EF, Scott Feldman5EFDOI: 10.12659/AJCR.927087
Am J Case Rep 2021; 22:e927087






Abstract
BACKGROUND: Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disease characterized by an intense immunologic response that results in multiorgan dysfunction. It typically manifests as a result of a familial genetic immunodeficiency disorder or secondary to a trigger such as an infection, malignancy, or autoimmune disease. The major factors involved in the development of the disease are an individual’s genetic propensity to develop HLH, such as rare associated mutations, or inflammatory processes that trigger the immune system to go haywire.
CASE REPORT: Before the COVID-19 pandemic, a 22-year-old woman with a history of congenital absence of the right kidney, right-sided hearing loss, and leukopenia presented with a 3-week history of generalized malaise, fever, chest pain, cough, and shortness of breath. She developed an acute systemic cytomegalovirus infection further complicated by HLH. Based on her history and clinical course, an underlying primary immunodeficiency was suspected. An immunodeficiency gene panel revealed a monoallelic mutation in GATA2, a gene that encodes zinc-transcription factors responsible for the regulation of hematopoiesis.
CONCLUSIONS: GATA2 deficiency encompasses a large variety of mutations in the GATA2 gene and leads to disorders associated with hematologic and immunologic manifestations of monocytopenia and B-, and natural killer-cell deficiency. Over time, affected individuals are at high risk of developing life-threatening infections and serious hematologic complications, such as myelodysplastic syndromes and/or leukemias. We aimed to illustrate the importance of identifying an underlying genetic disorder associated with secondary HLH to help guide acute and long-term management.
Keywords: GATA2 Transcription Factor, Lymphohistiocytosis, Hemophagocytic, GATA2 Deficiency, young adult
Background
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disease characterized by an intense immunologic response that results in multiorgan dysfunction. The underlying complex pathophysiology involves overactivation of macrophages and histiocytes, resulting in excessive cytokine release and the destruction of hematopoietic cells [1]. Studies based on animal models have found that HLH is predominantly driven by dysregulation and uncontrolled stimulation of CD8-positive T cells [2]. The North American Consortium for Histiocytosis (NACHO) classification of HLH syndrome distinguishes between “HLH disease” and “HLH disease mimics.” HLH syndrome broadly describes all genetic and environmental causes of pathologic immune dysregulation that meet the diagnostic criteria specified for HLH in 2004 (HLH-2004). HLH disease encompasses the following conditions: familial HLH with a distinct set of autosomal-recessive gene mutations of lymphocyte cytotoxicity in addition to HLH occurring secondary to malignancy, autoimmune disease, cytokine release syndrome, and immunocom-promise in patients who have either primary immunodeficiencies or treatment-related immune suppression. This includes patients with underlying primary immunodeficiency disorders who are particularly susceptible to infections or immunologic pathologies. HLH disease mimics refers to disorders that meet criteria for HLH but are caused by other conditions and will not benefit from immunosuppression, such as malignancy and certain infections. It is important to note that there is a substantial overlap between HLH disease and HLH disease mimics, particularly among subcategories of malignancy and immune compromise. The importance of the new categorization plays a role in treatment options for HLH and the utility of immunosuppression [2].
The major factors involved in the development of HLH are an individual’s genetic propensity to develop it, such as rare associated mutations, or inflammatory processes that trigger the immune system to go haywire. The most common external triggers leading to disease are infections, predominantly viruses such as cytomegalovirus (CMV), Epstein-Barr virus (EBV), and HIV [2]. Although the pathophysiology of HLH syndrome is not completely understood, it is believed that viruses provoke a hyperactive immune response in predisposed patients.
The clinical presentation of HLH is highly variable and follows a pattern of hyperinflammation involving multiple organ systems. Initial signs and symptoms include high fevers, splenomegaly, rash, and dyspnea in the setting of respiratory failure [3]. As defined by the 2004 HLH criteria, the diagnosis can be established if there is a verified molecular diagnosis consistent with HLH or 5 of the 8 clinical criteria are met. The clinical criteria are: fever; splenomegaly; cytopenia of more than 2 lineages in peripheral blood; hypertriglyceridemia and/or hypofibrinogenemia; hemophagocytosis in the bone marrow or spleen or lymph nodes; low or absent natural killer (NK)-cell activity; ferritin >500 ng/mL; and elevated soluble CD25 [4]. The diagnostic criteria are reflective of pathologic immune activation in HLH and are useful for assessing disease progression. Early recognition of the disease is critical because the risk of death is extremely high if treatment is delayed.
The heterogeneity of the disease processes in adults does not allow for a standardized treatment for HLH syndrome, and recommendations stem from the HLH-94 pediatric protocol. The management of adult HLH relies heavily on having a high index of suspicion for the disease and understanding the etiology or trigger for onset. An individualized approach is recommended, with the mainstays of therapy being immunosuppression and supportive care. Initiation of immunosuppressive treatment is recommended for patients who meet the clinical criteria and have severe disease. It is also crucial to consider diseases that mimic HLH and would not benefit from immunosuppression. Therapy for acutely ill patients should be in keeping with the HLH-94 protocol and includes induction with etopo-side and dexamethasone for 8 weeks. Further treatment options include emapalumab, an interferon-gamma-blocking antibody, in addition to dexamethasone in cases of recurrent or refractory disease [5]. Patients who do not exhibit severe disease with multiorgan dysfunction can be treated with steroids and observed for clinical improvement before starting immunosuppressive therapy. Supportive care includes transfusion and antimicrobial therapy for infection [2,5].
Case Report
INVESTIGATIONS:
The CTA was negative for pulmonary embolism; however, it revealed diffuse reticulonodular infiltrates in the lungs with numerous nodules, mediastinal lymphadenopathy, and splenomegaly (Figure 1). A bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial biopsy was performed and revealed mild hypervascularity of the airways without significant secretions. Samples were collected for cell count, culture, cytology, galactomannan assay, and flow cytometry. The cell count revealed a predominance of neutrophils. An echocardiogram demonstrated normal systolic function with an ejection fraction of roughly 60% to 65%. The blood cultures that were drawn on the patient’s admission showed no growth, and urine antigens for Streptococcus pneumoniae and Legionella pneumophila were negative. Multiple additional studies were obtained, including procalcitonin, viral serology for CMV and EBV, Aspergillus galactomannan, HIV, immunoglobulin (Ig), and an autoimmune panel. The results showed an elevated procalcitonin value of 4.58 ng/mL. Viral serology was significant for acute CMV infection with CMV IgM >240 AU/mL (reference range, <30 AU/mL) and CMV IgG 0.80 u/mL (reference range, <0.60 u/mL). CMV DNA quantification by PCR yielded a viral load of 1 890 000 IU/mL (reference range, undetected IU/mL). The serology results for EBV were highly suggestive of reactivation, with EBV capsid IgM Ag 46.60 U/mL (reference range, <36 U/mL), EBV capsid IgG Ag 553 U/mL (reference range, <18 U/mL), EBV early Ag IgG >150 U/mL (reference range, <9 U/mL), and EBV nuclear IgG Ag 49.6 U/mL (reference range, <18 U/mL). The EBV viral load detected was 60 540 copies/mL. A rheumatologic panel demonstrated an elevated antinuclear antibody count with a low titer of 1: 160 speckled pattern with ribonucleoprotein antibody 153 AU/mL (<100 AU/mL). It is important to note that an extensive autoimmune work-up was deferred until the patient’s acute illness had resolved. In addition, because she had no reported history of chronic symptoms, suspicion for lupus was low.
The patient’s Ig studies revealed normal levels of IgA (153 mg/dL; reference range, 70–400 mg/dL) and IgG (1000 mg/dL; reference range, 970–1,600 mg/dL) and elevated IgM (311; reference range, 40–230 mg/dL). The BAL cytology showed no malignant cells and the cultures were negative. The results of flow cytometry from the BAL sample could not be interpreted because not enough cells were collected. An abdominal ultrasound revealed increased echogenicity of portal triads, reflecting acute hepatitis, mild splenomegaly, and an absent right kidney. The patient’s spleen measured 13.5 cm longitudinally.
MANAGEMENT AND TREATMENT:
The patient was started on ganciclovir for treatment of an acute systemic CMV infection. Despite receiving broad-spectrum antimicrobial therapy and supportive care, she continued to deteriorate clinically and exhibited severe hypoxia, persistent fever, pancytopenia, and transaminitis. Additional laboratory studies were ordered to evaluate the patient’s anemia; the results revealed elevated levels of lactate dehydrogenase (LDH) (701 U/L; reference range, 85-240 U/L) and serum ferritin (27 514 ng/mL; reference range, 10–120 ng/mL). The patient’s serum fibrinogen level was elevated at 509 mg/dL and her triglycerides were 380 mg/dL. At this time, given the progression of the patient’s illness and the previously mentioned laboratory abnormalities, clinical suspicion was high for HLH. A bone marrow biopsy and aspiration were urgently performed. The biopsy revealed normocellular marrow with relative erythroid atypia, reactive plasmacytosis, and hemophagocytosis (Figure 2). The results of the bone marrow aspirate with flow cytometry and a myelodysplastic syndromes panel were negative.
The patient met 5 of 8 diagnostic criteria for the HLH-2004 protocol of fevers, splenomegaly, cytopenia, elevated ferritin, and bone marrow findings of hemophagocytosis. She was immediately started on intravenous (IV) Ig and dexamethasone 10 mg/m2. A transfer to another facility specializing in the HLH protocol was initiated. Supportive laboratory studies to aid diagnosis, including analysis of the interleukin-2 receptor, revealed that it was elevated at 2452 pg/mL.
Subsequent management of the patient focused on supportive care and antiviral therapy for acute systemic CMV infection complicated by the development of HLH. Because of concern about further immunosuppression with an active CMV infection, treatment with high-dose corticosteroids was discontinued. Despite ongoing treatment and a decreasing CMV viral load, the patient showed no significant improvement. Corticosteroids then were reintroduced (IV methylpredniso-lone, 40 mg/d). The patient subsequently began to rapidly improve, with higher blood counts and less hypoxemia. She ultimately did not receive etoposide because she began to show clinical improvement with steroids and treatment for CMV.
After extensive questioning and detailed history-taking throughout the patient’s hospital course, the patient recalled using more than the allotted sick days during high school and needing to be homeschooled. She reported having frequent upper respiratory viral infections, vaginal Candida infections, and severe acne. Based on the patient’s current hospitalization for CMV and HLH along with a history of recurrent viral and fungal infections, an underlying primary immunodeficiency disorder was suspected. Accordingly, specialized testing with immunologic and cytokine studies was performed, including IgG levels and lymphocyte subsets. A flow cytometric assessment of lymphocyte subsets for screening of primary immunodeficiency reflected low levels of B, CD4, and NK cells (Table 1). An NK-cell function test was inconclusive because the NK cells were negligible. A lymphocyte antigen mitogen panel demonstrated an absent lymphocyte response to Candida and tetanus, low response to phytohemagglutinin (PHA) and concanavalin A (Con A), and a normal response to pokeweed mitogen (PWM). Given the high probability that the patient had an underlying immune disorder, further work-up with whole exome sequencing was planned on an outpatient basis.
OUTCOME AND FOLLOW-UP:
During the patient’s hospitalization, the dynamics of her clinical symptoms correlated with progression of inflammation. She had nonspecific features of low-grade fever, fatigue, and shortness of breath that developed 3 weeks prior to her presentation. She then experienced additional symptoms of headache, poor appetite, and intermittent chest pain with palpitations. From Day 1 of her admission until about Day 3, she reported complaints of fever, chest pain, and cough. On Day 3, she suddenly developed acute shortness of breath, which worsened throughout her hospitalization. The severe dyspnea was associated with clinical findings of high fever, increased work to breathe, and tachycardia. The patient’s symptom course was consistent with an infectious picture; however, the distinguishing features that led to a diagnosis of HLH were the abnormalities on laboratory and imaging studies. They included transaminitis, elevated ferritin levels, and splenomegaly.
The patient eventually had significant improvement in hypoxia and was weaned off supplemental oxygen and onto room air. The empiric antibiotics she had been prescribed were eventually discontinued. After being hospitalized for 18 days, she was discharged for acute rehabilitation on a steroid taper and valganciclovir, 900 mg every 12 hr. As per the recommendation of the Infectious Disease specialists, valganciclovir was to be continued indefinitely until the patient’s CMV levels were undetectable. Approximately 2 months after discharge, the patient’s CMV viral load reached 2380 and she was continued on valganciclovir, 900 mg BID for suppressive therapy in the setting of a suspected primary immunodeficiency.
The patient ultimately underwent outpatient genetic testing with a primary immunodeficiency gene panel to assess 207 genes associated with inherited immune disorders. The panel identified a monoallelic mutation in the
Discussion
This case demonstrates an underlying primary immunodeficiency disorder, GATA2 deficiency, in a patient who presented with acute CMV-activated HLH [6]. The GATA2 protein is a transcription factor that regulates genes largely involved in hematopoietic and lymphatic stem cell maintenance. The disease is characterized by manifestations of monocytopenia and NK- and B-cell lymphopenia that predispose patients to viral, fungal, and mycobacterial infections. Syndromes commonly associated with GATA2 deficiency are monocytopenia and mycobacterial syndrome (MonoMAC); dendritic cell, monocyte, B, and NK lymphoid (DCML) deficiency; and primary lymphedema with myelodysplasia (Emberger syndrome) [4]. Importantly, depletion of or low NK-cell count is not only a part of GATA2 deficiency but also a diagnostic criterion for HLH. This strongly indicates a propensity for aggressive HLH.
The clinical presentation of GATA2 deficiency is widely variable, ranging from asymptomatic to severe, life-threatening conditions such as severe infections or hematologic malignancies. Patients are particularly susceptible to viral infections, most commonly EBV, varicella-zoster virus, and human papilloma-virus (HPV). Other manifestations include malignancies with increased risk for myeloid leukemia, autoimmune disorders, thrombotic events, lymphedema, and pulmonary alveolar proteinosis. An early diagnosis of GATA2 deficiency is vital for appropriate family screening, preventive care, and assessment of the role of transplantation [7,8]. Over time, patients are at high risk for developing myeloid leukemia, chronic myelomonocytic leukemia, and myelodysplasia. Long-term care must focus on frequent monitoring of peripheral blood cell counts and bone marrow cytogenetics to assess for potential progression to a hematologic pathology. In addition, prophylaxis with vaccinations for HPV and azithromycin for prophylaxis against myco-bacterial infections are the mainstays of long-term preventive care [8]. Allogeneic hematopoietic stem cell transplantation (HSCT) is the only curative treatment for GATA2 deficiency, given the progression in bone marrow failure [9].
Like other primary immunodeficiency disorders, GATA2 deficiency has been associated with the development of HLH syndrome [10]. Regardless of the classification and etiology of HLH, the presentation is defined by immune activation of the mononuclear phagocytic system, which leads to an exaggerated inflammatory response. Subsequent immune activation results in tissue damage and multiorgan failure. GATA2 deficiency is often missed as a diagnosis because of its rarity, variable clinical presentation, and lack of specific laboratory findings. Although bone marrow aspiration and biopsy are used as evidence to support diagnosis, findings of hemophagocytosis are neither sensitive nor specific for HLH. Commonly encountered findings in the bone marrow are phagocytosis of nucleated cells and activated macrophages [11]. Following confirmation of the diagnosis, rapid induction of dexamethasone and etoposide based on the HLH-94 protocol has resulted in survival rates of over 50% in pediatric patients with primary/familial HLH. The goal of therapy is to suppress inflammation and overactivation of immune cells. If the etiology of HLH is unknown, extensive work-up should include diagnostic studies to look for an underlying infectious process, malignancy, and/or autoimmune disorder. In all situations, treatment should not be delayed and should focus on halting the immune response [6]. In patients who do not respond to initial treatment, allogeneic HSCT should be considered. Before the use of SCT in primary HLH, nearly all patients relapsed and died [6,12].
A review of the literature shows that there have been only a handful of cases describing an underlying GATA2 deficiency in patients with acute secondary HLH [10]. Given the rarity of such a disease entity, treatment is highly variable. To better understand the correlation of GATA2 deficiency with the course of a widespread inflammatory response syndrome like HLH, we aimed to compare the approaches to management in a similar reported case. In a case described by Suzuki et al, the patient was started on corticosteroids with prednisolone 1 mg/kg/day before the etiology was known to be HLH [10]. Because of the progression of the illness, the patient received pulse-dose methylprednisolone (1 g/d for 5 days). The patient had clinical improvement on high-dose steroids and ultimately did not receive antiviral therapy once the diagnosis of CMV was established [10]. In comparison, our patient was treated for acute systemic CMV with antiviral drugs and high-dose steroids were administered once suspicion for HLH became high. It is important to note that induction therapy as outlined in the HLH protocol was not used in either case. Both of the patients had clinical improvement on high-dose steroid therapy, which suggests an underlying overwhelming immune activation characteristic of HLH. Although the treatments in the cases described differed, improvement in the patients’ illnesses likely can be attributed to the steroid therapy, which halted overactivation of the immune system, and a robust antibody response to CMV. In conclusion, the mainstay of therapy for HLH consists of treatment of the underlying cause, administration of immunosuppressive agents to blunt immune response, and supportive care.
Conclusions
This case provides insight on the importance of recognizing key elements and features in a patient with an acute viral syndrome triggered by HLH and an underlying primary immunodeficiency disorder. A key takeaway point to reiterate is the need to identify an underlying etiology, such as a primary immunodeficiency, in patients who have severe presentations in response to typical pathogens like CMV. Furthermore, the diagnosis of GATA2 deficiency in the setting of acute HLH warrants highly specialized care and prompt evaluation for HSCT. Because such care requires an individualized approach with detailed management, treatment in centers with highly trained specialists is important to optimize patient outcome. In addition, timely diagnosis is essential to lower the risks of morbidity and mortality in all patients who are suspected of having HLH syndrome.
Figures
References:
1.. Allen CE, Yu X, Kozinetz CA, Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis: Pediatr Blood Cancer, 2008; 50; 1227-35
2.. Jordan MB, Allen CE, Greenberg J, Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the North American Consortium for Histiocytosis (NACHO): Pediatr Blood Cancer, 2019; 66(11); e27929
3.. Nikiforow S, Berliner N, The unique aspects of presentation and diagnosis of hemophagocytic lymphohistiocytosis in adults: Hematology Am Soc Hematol Educ Program, 2015; 2015(1); 183-89
4.. Bode SF, Ammann S, Al-Herz W, The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: Implications for differential diagnosis and pathogenesis: Haematologica, 2015; 100; 978-88
5.. La Rosée P, Horne A, Hines M, Recommendations for the management of hemophagocytic lymphohistiocytosis in adults: Blood, 2019; 133(23); 2465-77
6.. Atim-Oluk M, Cytomegalovirus associated haemophagocytic lymphohistiocytosis in the immunocompetent adult managed according to HLH-2004 diagnostic using clinical and serological means only: Eur J Microbiol Immunol, 2013; 3; 81-89
7.. Spinner M, Ker JP, Stoudenmire CJ, GATA2 deficiency underlying severe blastomycosis and fatal herpes simplex virus-associated hemophagocytic lymphohistiocytosis: J Allergy Clin Immunol, 2016; 137; 638-40
8.. Hsu A, McReynolds L, Holland S, GATA2 deficiency: Curr Opin Allergy Clin Immunol, 2015; 15; 104-9
9.. Parta M, Shah NN, Baird K, Allogeneic hematopoietic stem cell transplantation for GATA2 Deficiency using a busulfan-based regimen: Biol Blood Marrow Transplant, 2018; 24; 1250-59
10.. Suzuki T, Takaya S, Kunimatsu J, GATA2 mutation underlies hemophagocytic lymphohistiocytosis in an adult with primary cytomegalovirus infection: J Infect Chemother, 2020; 26; 252-56
11.. Reiner AP, Spivak JL, Hematophagic histiocytosis. A report of 23 new patients and a review of the literature: Medicine (Baltimore), 1988; 67; 369-88
12.. McClain K, Eckstein O: Treatment and prognosis of hemophagocytic lymphohistiocytosis, 2014, Waltham, MA, UpToDate
Figures
Tables
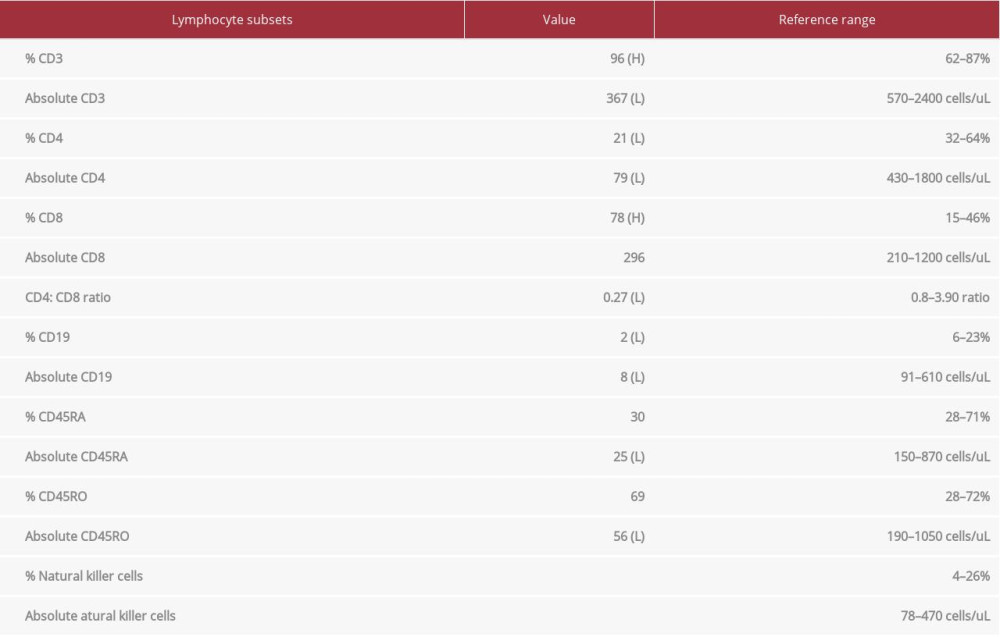 Table 1.. Flow cytometric screening of lymphocyte subsets for primary immunodeficiency. The results revealed low levels of B, CD4s, and natural killer cells.Table 1.. Flow cytometric screening of lymphocyte subsets for primary immunodeficiency. The results revealed low levels of B, CD4s, and natural killer cells.
Table 1.. Flow cytometric screening of lymphocyte subsets for primary immunodeficiency. The results revealed low levels of B, CD4s, and natural killer cells.Table 1.. Flow cytometric screening of lymphocyte subsets for primary immunodeficiency. The results revealed low levels of B, CD4s, and natural killer cells. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953068
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133