17 February 2021: Articles 
Idiopathic Acquired Hemophilia A, a Rare Cause of Bleeding: A Case Report and Literature Review
Challenging differential diagnosis, Diagnostic / therapeutic accidents, Management of emergency care, Rare disease, Educational Purpose (only if useful for a systematic review or synthesis)
Carlos Andrés Regino1ABCDEFG*, José C. Alvarez1ABCDEFG, Leonardo Mejía Buriticá2ABCDEFG, Natalí Uribe Pulido1ABCDEF, Valeria Torres Yepes3ABCDEFG, José D. Torres24ABCDEFGDOI: 10.12659/AJCR.929401
Am J Case Rep 2021; 22:e929401






Abstract
BACKGROUND: Acquired hemophilia is a bleeding disorder mediated by an autoimmune process, in which antibodies against clotting factors are developed. This is a rarely suspected complex condition in which the initial manifestations are spontaneous bleeding in the skin, soft tissues, and mucosa in patients with no known history of bleeding disorders. Most of the cases are idiopathic (50%), but it can be associated with autoimmune diseases, malignancy, pregnancy, and medications. The most frequent type is mediated by inhibitors against factor VIII, followed by coagulation factor IX and XI. It is a disease with high morbidity and mortality rates without adequate treatment. Diagnosis is based on the detection of low concentrations of clotting factors and the presence of an inhibitor.
CASE REPORT: We present 2 cases of patients with spontaneous bleeding in whom the diagnosis of idiopathic acquired hemophilia A was made, an extensive malignancy study was performed that was negative, and the presence of autoimmunity markers (positive antinuclear antibodies (ANA)) was observed, without any another sign of autoimmune disease. They received immunosuppressive therapy with bleeding control and inhibitor eradication.
CONCLUSIONS: Acquired hemophilia A is a rare but potentially lethal disease, representing a medical challenge from its diagnosis to its treatment. An early recognition and treatment are fundamental because delays are associated with adverse outcomes. Optimal management includes the workup and treatment for an underlying disease, use of “bypass” agents when active bleeding presents, and inhibitor titer eradication through immunosuppressants drugs. With the present cases, we highlight the importance of considering acquired hemophilia A in older patients with similar symptoms, to achieve early diagnosis and treatment.
Keywords: Autoimmunity, Blood Coagulation Disorders, Hemophilia A, Autoimmune Diseases, Hemorrhage, Immunosuppressive Agents, Pregnancy
Background
Acquired hemophilia A (AHA) is a hemorrhagic disorder mediated by an autoimmune process, in which antibodies are developed against clotting factors [1]. Such disorder alters hemostatic functions, and presents with hemorrhagic signs that can be life threatening. It is characterized by sudden bleeding in skin, mucosal bleeding, and soft tissue bleeding in absence of personal or family history of clotting disorders. Herein, we present 2 cases with acquired hemophilia A.
Case Reports
CASE 1:
A 60-year-old woman with personal history of active smoking and arterial hypertension treated with losartan presented to the emergency room (ER) for 1 month of spontaneous ecchymoses appearing in the upper limbs. Two weeks before the ER consult, the patient presented with subconjunctival hemorrhage, hematomas, and ecchymoses in the thighs (Figure 1), without pain or functional limitation.
Among laboratory findings, she presented a normocytic anemia (hemoglobin [Hb] 8.2 g/dL, mean corpuscular volume [MCV] 90 fL) with normal leucocytes and platelets. Creatinine was 0.67 mg/dL, and infection workup was negative (HIV, hepatitis B and C virus). Partial thromboplastin time (PTT) was prolonged in 58.2” with a normal prothrombin time (PT) of 12.2”.
Mixing tests with a 2-hour incubation at a 37°C temperature did not correct with normal plasma, factor VIII level was 1.5%, and Bethesda assay with 80 units confirmed the presence of an inhibitor (Table 1).
A diagnosis of acquired idiopathic hemophilia A was established. The presence of autoimmunity markers was observed (antinuclear antibodies [ANA] titer 1: 160, homogeneous pattern), without any other sign of autoimmune disease. Treatment with prednisone at 1 mg/kg/day was started at 1 month, then tapered. The bleeding signs were controlled and the inhibitor was eradicated.
CASE 2:
A 73-year-old woman with personal history of surgically corrected scoliosis and active smoking consulted to the ER for an extensive hematoma of the upper left limb, which appeared 15 days after a venous sample was taken for routine laboratory tests. She then developed multiple sudden ecchymoses in the chest, upper limbs, and lower right limb (Figures 2, 3). She had no fever or constitutional symptoms, her vital signs were normal, and the rest of the physical examination was unremarkable.
Among laboratory findings, she had normocytic anemia (Hb 8.7 g/dL; MCV 84 fL), with normal leucocytes and platelets. Creatinine was 0.7 mg/dL, with a normal urinalysis and urinary sediment. Infection workup was negative for HIV and hepatitis B and C viruses. The PTT was prolonged at 126” and the PT was normal at 11.9”. Mixing tests did not correct with normal plasma, even after a 37°C incubation was performed, the factor VIII level was 1.58%, and the Bethesda assay showed levels of 9.7 UB. An extensive workup was performed for malignancy, which was negative. ANA were positive at a 1: 80 titer with a cytoplasmic pattern. Extractable nuclear antigens and rheumatoid factor were negative (Table 2).
Therefore, a diagnosis of acquired idiopathic hemophilia A was made. During the inpatient stay, she presented with left shoulder hemarthrosis and her hematomas increased in size, for which she was treated with recombinant factor VII, with improvement of her symptoms. Prednisone treatment was started at a dose of 1 mg/kg/day and cyclophosphamide at a dose of 1 mg/kg/day, with good clinical evolution and eradication of inhibitor titers.
Discussion
Acquired hemophilia A is characterized by spontaneous production of antibodies against endogenous factor VIII [2]. Most antibodies are polyclonal IgG, especially subtype IgG4, that neutralize the clotting action of the factor [3]. These antibodies bind to C2 domain of factor VIII. Loss of C2 domain, which binds to phosphatidylserine in activated platelets, endothelial cells, and von Willebrand factor, leads to a reduced clotting activity (Figure 4) [4,5].
It is a rare autoimmune disorder, with an annual incidence of 1.5 cases per million [6,7], and is grossly underestimated as most cases are diagnosed within fatal complications. AHA is a disease of elderly patients (mean age 64–78 years), but can be associated with pregnancy or autoimmune diseases, as it has been previously documented in cohorts with younger patients. The incidence in men and women is similar, with an exception in the group age of 20–40 years due to pregnancy-related cases [7].
Most cases are idiopathic (43.6–51.9%), but some cases are related to malignancy (6.4–18.4%), autoimmune disorders (9.4–17%), infections, cutaneous diseases, and drugs [6]. These conditions associated with AHA are summarized in Table 3.
In a recent revision of 105 cases of AHA associated with malignancy, 60 patients had solid tumors and 45 patients had a hematologic malignancy; the most common solid tumors were: prostate cancer (25.3%), lung cancer (15.8%), and colon cancer (9.5%). The most common hematologic malignancies were: lymphoma (24.4%), chronic lymphocytic leukemia (22.3%), plasmatic cells dyscrasias (20%), chronic myeloproliferative malignancies (13.3%), acute myeloid leukemia (9%), myelodys-plastic syndrome (8.8%), and mycosis fungoid (2.2%) [8]. In comparison to idiopathic AHA patients, cancer-related AHA patients are more likely to have recurrent bleeding, and less likely to achieve complete response to inhibitor eradication [8,9].
In general, AHA patients present with subcutaneous hematomas (80%) in the following locations: intramuscular (45%), gastrointestinal (21%), genitourinary (9%), and retroperitoneal (9%) [2,10]. Unlike congenital hemophilias, hemarthrosis is rare in AHA [11]. The fatal bleeding rate may be as high as 8%, mainly with intracranial and deep-tissue bleeds [10].
The diagnosis of AHA is considered a challenge, given its non-specific presentation and its low prevalence. More than 95% of patients are symptomatic at the time of diagnosis. The coagulation test shows an isolated prolonged PTT, with a normal PT, a reduced activity of factor VIII (<1% in 50% of cases, <5% in 75% of cases, <40% in 100% of cases) [12], and the presence of a clotting factor inhibitor [13]. Quantitative measurement of clotting factors activity and the inhibitor’s level is considered a crucial diagnostic step. The quantification of the specific inhibitor is performed through the modified Bethesda assay [2].
Nonetheless, there is no correlation between the bleeding severity and the FVIII levels or the inhibitor titer [14]. Therefore, these measurements should not guide treatment decisions.
The PTT is prolonged due to an intrinsic pathway clotting factor deficiency, or the presence of an inhibitor against one of these factors: autoantibodies, lupus anticoagulant, or pharmacologic anticoagulants [15]. The mixing tests differentiate a clotting factor deficiency from the presence of an inhibitor [1].
Acquired antibodies against FVIII have a complex kinetics as they are both time- and temperature-dependent; therefore, the PTT results have to be compared as follows: 1) obtained immediately after mixing normal and patient’s plasma and 2) after a 2-hour incubation [16]. An abnormal prolongation of PTT of the mixing test after the incubation is typical of auto-antibodies against FVIII. Prolonged PTT values from the 1: 1 mixing test (normal plasma and patient’s plasma), which are similar at baseline and after incubation, are suggestive of lupus anticoagulant, but it has to be confirmed with the dilute Russell’s viper venom time (dRVVT). Moreover, clinical presentation aids in differential diagnosis, as AHA patients usually have a bleeding phenotype, contrary to patients with a lupus anticoagulant [1]. However, it is worth remarking that AHA and lupus anticoagulant are autoimmune disorders that can coexist in the same patient [17,18].
Treatment is focused in controlling and preventing bleeding, inhibitor eradication, and the management of the underlying disease in secondary cases. Treatment decisions are usually based on clinical experience, as there is no robust evidence available. However, international guidelines have been published about the management of patients with AHA [2,30]. The general treatment recommendations are summarized in Table 4.
To achieve the first hemostatic target, recombinant activated clotting factor VII, activated prothrombin complex concentrate (APCC), and porcine recombinant factor VIII are used. There are no clear advantages in efficacy or safety among these, and any can be used as a first-line therapy [18–20].
It is important to note that, independent of inhibitor titer, in patients with active bleeding, the first-line therapy must be any of the bypass agents; the FVIII replacement at high doses and desmopressin is usually reserved for cases where bypass agents are not available [1].
Desmopressin, which stimulates the release of FVIII and VWF from endothelial cells and can provide a transient rise in FVIII levels, may also be effective in AHA [21]. A systematic review of the literature was published, with 37 patients collected from 15 case reports where desmopressin was used for the treatment of non-life-threatening hemorrhages or to cover minor surgical, invasive, or dental procedures. The median inhibitor titer was 5.3 BU (range, 0-444 BU), median FVIII level prior to desmopressin administration was 5% (range, 0–40%), and hemostatic response was achieved in 57%, with an approximately 5-fold increase in FVIII level following desmopressin administration [22]. Similar results were observed by Mudad and Kane [23], in which the best responses (19/20 cases [95%]) were observed in patients with basal FVIII levels higher than 5%.
The literature supports the role of desmopressin in the treatment of minor bleeding episodes or prophylaxis of minor surgical/invasive procedures in patients with acquired hemophilia A with a low inhibitor titer (<5 BU) and baseline levels of FVIII> 5%. Nevertheless, experience in this setting is limited to a few case reports [22] and analysis of EACH2 showed that the efficacy of human FVIII concentrates and desmopressin was clearly lower than that of bypassing agents [1].
For inhibitor eradication, which is important to reduce the long-term lethal bleeding risk, immunosuppressants are used: steroids alone or in combination with cyclophosphamide or rituximab [1].
The United Kingdom Vigilance study defined complete remission as: normal FVIII and indetectable inhibitor with suspension of immunosuppression, or the lower dose that maintains response [10]. The taskforce of the German, Austrian, and Swiss Society of Thrombosis and Hemostasis (GTH) also included a definition of partial remission: FVIII >50% and no bleeding after hemostatic treatment suspension for at least 24 hours [12]. According to GTH data, FVIII level <1% or an inhibitor titer >20 UB are predictors of poor response; hence, an initial combined therapy is recommended. Recently, based on an FVIII/VWF antigen (VWF: Ag) ratio (FVIII/W ratio) close to 1.0, an observational study determined throughout the first weeks of immunosuppression, the FVIII/W ratio gradually increased and stayed above 0.66 in all patients who did not relapse, suggesting this biological marker could be considered to predict recovery and/or relapse in AHA [24]. In another study, neither FVIII >0.5 IU/mL nor FVIII inhibitor titers <0.6 UB were potential risk factors for relapse, but it was associated with older age, male sex, and lymphoproliferative syndromes, although relapse is not linked to worse overall survival [25].
There is an increasing experience with rituximab. A systematic review of 65 AHA patients treated with rituximab only or in combination with other immunosuppressants observed a complete or partial response in more than 90% of patients (59/65 cases), being the presence of high inhibitor titers (>100 UB/mL), which a negative predictive factor for rituximab response [26]. In the EACH2 registry, 30 of 51 patients (59%) treated with an immunosuppressant regimen including rituximab achieved stable response, with a success rate half of those regimens with steroids only (48%) or steroids and cyclophosphamide (70%). Seven of 14 patients that relapsed after steroids as a first-line therapy achieved a stable response with regimens based on rituximab. Current recommendations support the use of rituximab as a first-line therapy in AHA patients with negative predictive factors, resistant to steroids, or relapsing disease after steroid treatment [12].
Emicizumab, a new monoclonal bispecific antibody that contains 2 binding fragments to clotting factors IX and X, was developed for the treatment of hereditary hemophilia with inhibitors, and it has been used in patients with AHA given the similar pathophysiology [27,28]. In a clinical trial of 12 patients with AHA, FVIII <1%, and a mean inhibitor titer of 22.3 UB/ mL (range 3-2000), hemostatic efficacy was achieved within 3 days after the first dose was administered. Severe bleeding stopped without any increase in adverse events. The administration of emicizumab seems to be an important novel therapy for hemostasis in AHA patients, with the potential of preventing bleeding, reducing secondary effects, reducing costs, and protecting patients from thromboembolic complications [29].
Given the risks of adverse effects with immunosuppressant therapy and the risk of recurrence, which has been reported in up to 23% of cases [21], patients must undergo FVIII level measurement once the clinical response is achieved, monthly first, and then the time interval can be broadened when response holds up [1].
In our patients the cause of the acquired hemophilia was not determined, but a sustained remission of the disease was obtained by using immunosuppressive treatment based on prednisone alone in the first case and combined with cyclophosphamide in the second.
Conclusions
Acquired hemophilia A is a rare but potentially lethal disease, representing a medical challenge from its diagnosis to its treatment. Early recognition and treatment are fundamental because delays are associated with adverse outcomes. Optimal management includes workup and treatment for an underlying disease, use of “bypass” agents when active bleeding presents, and inhibitor titer eradication through immunosuppressant drugs. With the present cases, we highlight the importance of considering acquired hemophilia A in older patients with similar symptoms, to achieve early diagnosis and treatment.
Figures
References:
1.. Tiede A, Collins P, Knoebl P, International recommendations on the diagnosis hemophiliaand a treatment of acquired: Haematologica, 2020; 105(7); 1791-801
2.. Kruse-Jarres R, Kempton CL, Baudo F, Acquired hemophilia A. Updated review of evidence and treatment guidance: Am J Hematol, 2017; 92(7); 695-705
3.. Viesca-Contreras V, Amatón-Tabares R, Duque-Rodríguez J, Hemofilia adquirida tipo A. Reporte de caso: Revista Medica, 2017; 8(04); 113-18 [in Spanish]
4.. Arai M, Scandella D, Hoyer LW, Molecular basis of factor VIII inhibition by human antibodies. Antibodies that bind to the factor VIII light chan prevent the interaction of factor VIII with phospholipid: J Clin Invest, 1989; 83(6); 1978-84
5.. Franchini M, Gandini G, Di Paolantonio T, Mariani G, Acquired hemophilia A: A concise review: Am J Hematol, 2005; 80(1); 55-63
6.. Franchini M, Mannucci PM, Acquired haemophilia A: A 2013 update: Thromb Haemost, 2013; 110(6); 1114-20
7.. Sborov DW, Rodgers GM, Acquired hemophilia A: A current review of autoantibody disease: Clin Adv Hematol Oncol, 2012; 10(1); 19-27
8.. Napolitano M, Siragusa S, Mancuso S, Kessler CM, Acquired haemophilia in cancer: A systematic and critical literature review: Haemophilia, 2018; 24(1); 43-56
9.. Saito M, Ogasawara R, Izumiyama K, Acquired hemophilia A in solid cancer: Two case reports and review of the literature: World J Clin Cases, 2018; 6(14); 781-85
10.. Collins PW, Hirsch S, Baglin TP, Acquired hemophilia A in the United Kingdom: A 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ organisation: Blood, 2007; 109(5); 1870-77
11.. Mingot-Castellano ME, Núñez R, Rodríguez-Martorell FJ, Acquired haemophilia: Epidemiology, clinical presentation, diagnosis and treatment: Med Clin (Barc), 2017; 148(7); 314-22
12.. Tiede A, Klamroth R, Scharf RE, Prognostic factors for remission of and survival in acquired hemophilia A (AHA):Results from the GTH-AH 01/2010 study: Blood, 2015; 125(7); 1091-97
13.. Casas Patarroyo CP, Agudelo López CDP, Galvez K, [Adequate diagnosis of acquired hemophilia A]: Rev Med Chil, 2019; 147(3); 334-41 [in Spanish]
14.. Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A, Acquired haemophilia: Review and meta-analysis focused on therapy and prognostic factors: Br J Haematol, 2003; 121(1); 21-35
15.. Lossing TS, Kasper CK, Feinstein DI, Detection of factor VIII inhibitors with the partial thromboplastin time: Blood, 1977; 49(5); 793-97
16.. Tiede A, Werwitzke S, Scharf RE, Laboratory diagnosis of acquired hemophilia A: limitations, consequences, and challenges: Semin Thromb Hemost, 2014; 40(7); 803-11
17.. Seethala S, Collins NP, Comerci G, An unusual etiology for elevation of activated partial thromboplastin time (aPTT) in SLE: Acquired hemophilia and lupus anticoagulant: Case Rep Hematol, 2013; 2013; 521785
18.. Baudo F, Collins P, Huth-Kühne A, Management of bleeding in acquired hemophilia A: Results from the European Acquired Haemophilia (EACH2) registry: Blood, 2012; 120(1); 39-46
19.. Kruse-Jarres R, St-Louis J, Greist A, Efficacy and safety of OBI-1, an antihaemophilic factor VIII (recombinant), porcine sequence, in subjects with acquired haemophilia A: Haemophilia, 2015; 21(2); 162-70
20.. Tiede A, Worster A, Lessons from a systematic literature review of the effectiveness of recombinant factor VIIa in acquired haemophilia [published erratum appears in Ann Hematol, 2018, 97(10): 1889–1901]: Ann Hematol, 2018; 97(12); 2531
21.. Mannucci PM, Hemostatic drugs: N Engl J Med, 1998; 339; 245-53
22.. Collins PW, Management of acquired haemophilia A: J Thromb Haemost, 2011; 9(Suppl. 1); 226-35
23.. Mudad R, Kane WH, DDAVP in acquired hemophilia A: Case report and review of the literature: Am J Hematol, 1993; 43; 295-99
24.. Trossaert M, Graveleau J, Thiercelin-Legrand MF, Ag ratio as a useful tool to predict relapse in patients with acquired haemophilia A: A retrospective cohort study: Haemophilia, 2019; 25(3); 527-34
25.. Mizrahi T, Doyon K, Dubé E, Relapse pattern and long-term outcomes in subjects with acquired haemophilia A: Haemophilia, 2019; 25(2); 252-57
26.. Franchini M, Rituximab in the treatment of adult acquired hemophilia A: A systematic review: Crit Rev Oncol Hematol, 2007; 63(1); 47-52
27.. Lenting PJ, Denis CV, Christophe OD, Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: How does it actually compare to factor VIII?: Blood, 2017; 130(23); 2463-68
28.. Oldenburg J, Mahlangu JN, Kim B, Emicizumab prophylaxis in hemophilia A with inhibitors: N Engl J Med, 2017; 377(9); 809-18
29.. Knoebl P, Thaler J, Jilma P, Emicizumab for the treatment of acquired hemophilia A: Blood, 2020 [Online ahead of print]
30.. Huth-Kühne A, Baudo F, Collins P, International recommendations on the diagnosis and treatment of patients with Acquired hemophilia A: Haematologica, 2009; 94(4); 566-75
Figures
Tables
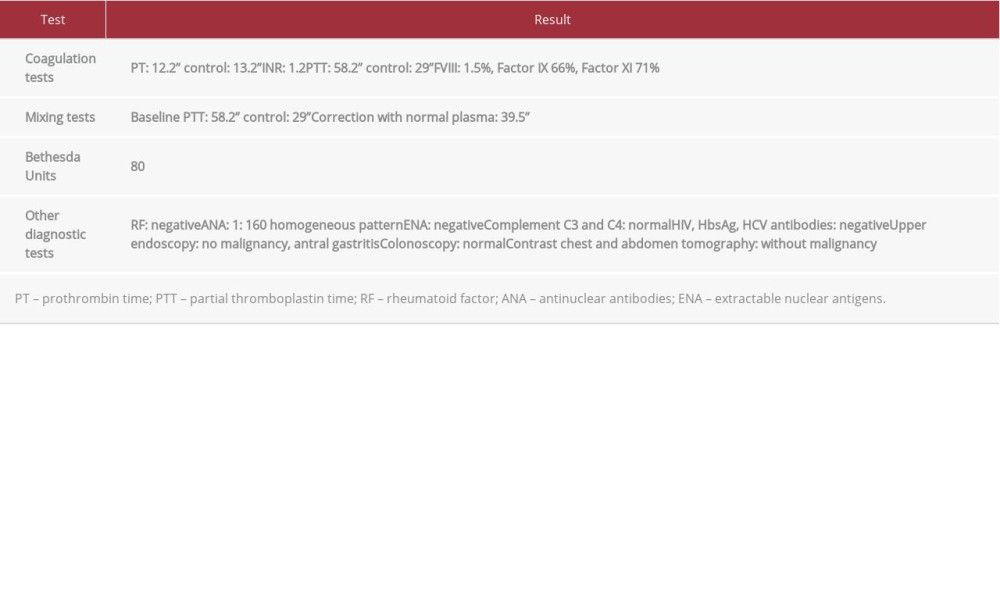 Table 1.. Laboratory findings of case 1.
Table 1.. Laboratory findings of case 1.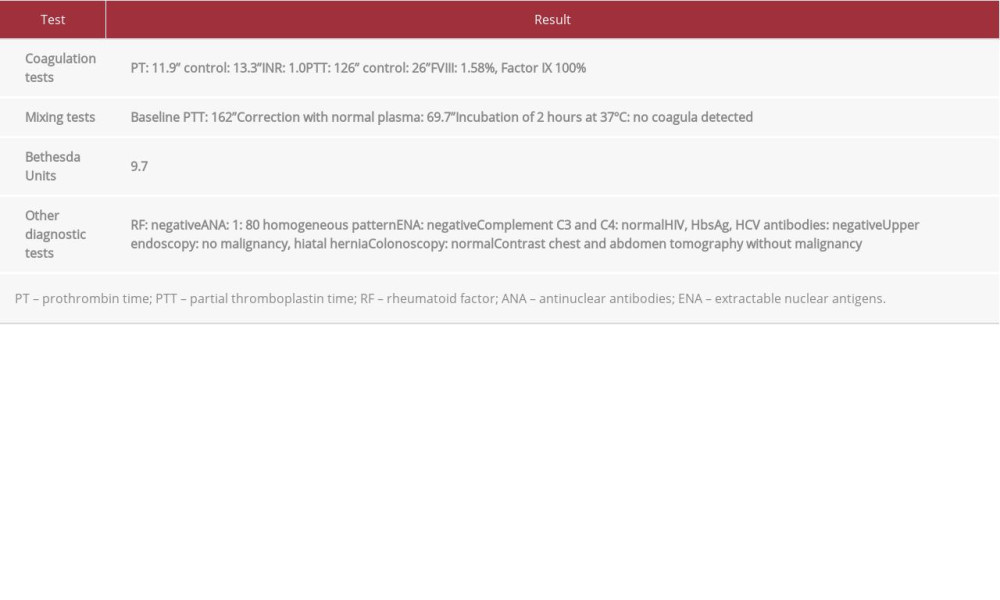 Table 2.. Laboratory findings of case 2.
Table 2.. Laboratory findings of case 2.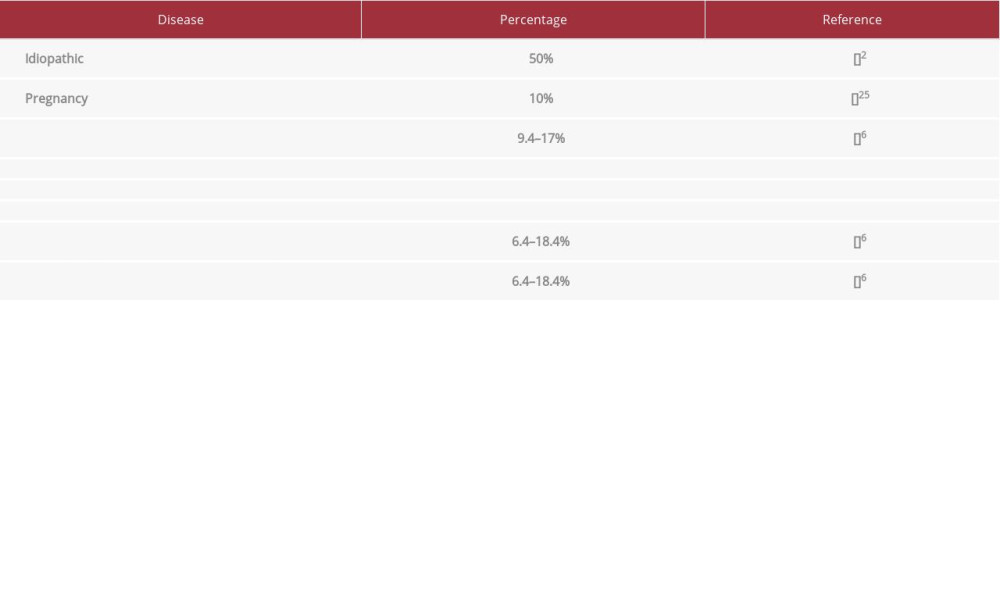 Table 3.. Diseases associated with acquired hemophilia.
Table 3.. Diseases associated with acquired hemophilia.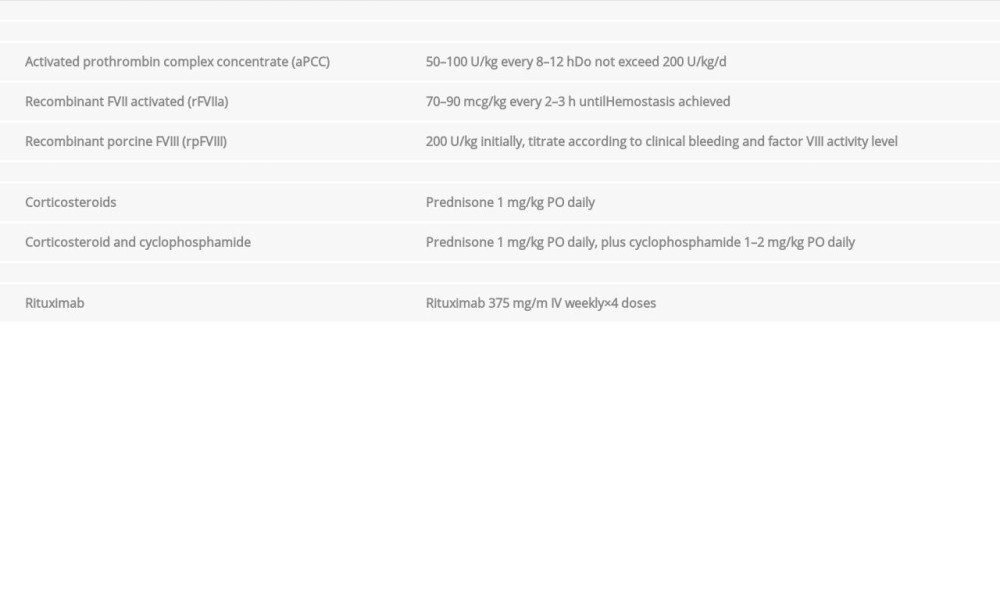 Table 4.. Guidelines recommendations for treatment of AHA. Modified of references [2,30].Table 1.. Laboratory findings of case 1.Table 2.. Laboratory findings of case 2.Table 3.. Diseases associated with acquired hemophilia.Table 4.. Guidelines recommendations for treatment of AHA. Modified of references [2,30].
Table 4.. Guidelines recommendations for treatment of AHA. Modified of references [2,30].Table 1.. Laboratory findings of case 1.Table 2.. Laboratory findings of case 2.Table 3.. Diseases associated with acquired hemophilia.Table 4.. Guidelines recommendations for treatment of AHA. Modified of references [2,30]. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report  22,759,844
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133