30 September 2021: Articles 
Hemophagocytic Lymphohistiocytosis Secondary to Peripheral T Cell Lymphoma with Rapid Onset and Fatal Progression in a Young Patient: A Case Report and Review of the Literature
Challenging differential diagnosis, Rare disease
Patrycja Paluszkiewicz1CDEF, Adrian Martuszewski2CDEF, Maciej Majcherek3ABCDE*, Marta Kucharska4BC, Aleksandra Bogucka-Fedorczuk3CDE, Tomasz Wróbel3EFG, Anna Czyż3ABCDEFDOI: 10.12659/AJCR.932765
Am J Case Rep 2021; 22:e932765






Abstract
BACKGROUND: Constant stimulation of lymphocytes and histiocytes can result in hemophagocytic lymphohistiocytosis (HLH), which can be primary or secondary (sHLH). The main causes of sHLH are infections and hematological malignancies, especially non-Hodgkin lymphoma. Despite new insights into the pathogenesis of HLH, the diagnosis and treatment of this immune disorder remain a great challenge.
CASE REPORT: We present a case of a young adult without comorbidities whose clinical course was nonspecific for several months and resulted in late diagnosis of HLH secondary to peripheral T cell lymphoma (PTCL). The etiological factor of recurring fever, hepatosplenomegaly, and deteriorating condition was unidentified for a long time before fatal sHLH was finally diagnosed. The patient was treated according to the HLH-2004 protocol; however, he did not achieve any response. Unfortunately, due to nonspecific symptoms, lack of lymphadenopathy for a long time, and negative positron emission tomography results, the diagnosis of PTCL was established only after the patient’s death.
CONCLUSIONS: It should be emphasized that early diagnosis is crucial for better prognosis of patients with sHLH. Bone marrow biopsy is worth considering in patients with prolonged fever of unknown origin, hyperferritinemia, splenomegaly, and unexplained cytopenia of 2 or more lineages. Despite the existence of diagnostic and therapeutic protocols available in the literature, the prompt diagnosis and treatment of HLH remains a great challenge. More precise and specific diagnostic tools for HLH are needed.
Keywords: Fever of Unknown Origin, Lymphohistiocytosis, Hemophagocytic, Lymphoma, T-Cell, Peripheral, Sepsis, Bone Marrow, Fever, Humans, young adult
Background
Hemophagocytic lymphohistiocytosis (HLH) was described for the first time in 1939 by Scott in The Lancet [1]. The authors named the disease histiocytic medullary reticulosis to replace the diagnosis of atypical Hodgkin’s disease. All described cases ended with the death of the patient, and the post-mortem examination showed proliferation of erythrophagocytic histiocytes and their precursors throughout the lymphoreticular tissue. The cases were described in terms of histopathological and symptomatic aspects without any specific treatment of the patients.
The first description of the familial hemophagocytic lymphohistiocytosis (FHL) was published in 1952, in which a series of cases in children was presented. It was then proposed to name the disease hemophagocytic reticulosis. In the first patient, as reported by researchers, a fever resistant to massive doses of penicillin occurred and transfusion was performed. In the second patient, adrenocorticotrophic hormone therapy was used mainly because of hypersplenism, which resulted in regressing of hypersplenism and an increase of hemoglobin level. The patient died after a second admission. He presented with pallor and vomiting, followed by fever and skin sunburnt, without bilirubin serum level increase [2].
In the light of current knowledge, HLH is a clinical disorder caused by an excessive inflammatory reaction to the initiating event. These initiating events cause constant stimulation of lymphocytes and histiocytes, as a result of which, excessive amounts of proinflammatory cytokines are secreted. Cytokine reaction gradually increases and ineffectively tries to stimulate cells of the immune system. In fact, the dysregulation of the secretion of proinflammatory cytokines derived from lymphocytes such as IL-2, INF-gamma, and cytokines derived from macrophages such as IL-1b, TNF alpha, IL-6, IL-18, as well as the Toll-like receptor 9 (TLR-9) plays a key role in the pathogenesis of HLH. In addition, increased levels of regulatory cytokines, cytokine inhibitors (soluble TNF receptor, IL-1b receptor antagonist) are also observed [3]. Finally, a “cytokine storm” leads to excessive macrophage activation and uncontrolled hemophagocytosis, which in turn causes severe cytopenia. These disorders of the immune system are reflected in the HLH clinical presentation: fever, enlargement of liver and spleen, cytopenia, and neurological abnormalities [4,5].
Hemophagocytic lymphohistiocytosis might be primary, referred to as FHL, or secondary (secondary hemophagocytic syndrome, secondary HLH, sHLH) [5,6]. The symptoms of primary HLH usually occur within the first months or years of life, but in some patients with primary HLH the first symptoms appear later, in adolescence or early adulthood and are called late-onset primary HLH. The onset of symptoms may be related to initiating factors similar to those in secondary HLH [7]. In FHL, autosomal recessive inheritance can be detected at birth with screening tests (screen for genetic HLH diseases) and treated with relatively good outcomes, which has improved the prognosis. Unfortunately, these screening tests are not available worldwide. However, FLH is still an extremely dangerous disease, for which a bone marrow transplant is required. It is caused by a congenital failure of natural killer (NK) cells and cytotoxic T cells [8]. Secondary HLH mostly follows Epstein-Barr virus (EBV) infection, but may be also induced by malignancies (known as malignancy-associated HLH or M-HLH). Non-Hodgkin lymphomas (NHL) are a risk factor for sHLH, with a reported prevalence of HLH up to 20% in some subtypes. According to the 2016 World Health Organization (WHO) classification, this group of lymphoid neoplasms includes peripheral T cell lymphomas (PTCL), which is more aggressive in their natural behavior than is B cell NHL [9,10]. Among the subtypes of lymphoma-associated hemophagocytic syndrome (LAHS), the most common (35%) is NK/T nasal type-LAHS [11]. Among the LAHS subtypes, NK/T-LAHS has an extremely poor prognosis, with a 5-year patient survival below 15% [12]. A single-center retrospective Swedish study showed that the estimated 1-year incidence of M-HLH in adults is 0.36/100 000 individuals/year and is always associated with poor outcome [13].
Of note, sHLH related to COVID-19 is a new challenge [14]. Eroglu et al suggested that all patients with COVID-19 should be monitored for HLH, but obviously not all patients with hyperinflammation meet the criteria for HLH [15]. Parasites and fungi are a less common cause of HLH, with histoplasma, leishmania, plasmodium, and toxoplasma infections being the most common [16].
HLH can develop in the course of rheumatic diseases as a potentially life-threatening systemic hyperinflammatory disorder, termed macrophage activation syndrome (MAS) by rheumatologists [17,18]. It occurs mainly in patients with Still disease or juvenile idiopathic arthritis (JIA), but also occurs in the course of systemic lupus erythematosus (SLE) [19].
Diagnosis and treatment of HLH remain a great challenge. In daily clinical practice, the clinical and laboratory criteria of HLH94 and HLH04 are usually used.
These criteria include laboratory and clinical features: (1) fever, (2) splenomegaly, (3) bicytopenia, (4) hypertriglyceridemia and/or hypofibrinogenemia, (5) hyperferritinemia, (6) hemophagocytosis in bone marrow or spleen or lymph nodes, (7) low or absent NK cell activity, and (8) soluble CD25 higher than 2400 U/ml. If hyperferritinemia is at least 2000 μg/l, fulfillment of the 4 clinical criteria can be considered sufficient. Indeed, hyperferritinemia exceeding 70 000 μg/l in some cases is the most characteristic laboratory feature for HLH [20,21].
However, widespread use of the HLH04 criteria is hampered by the fact that they have been validated in children with primary HLH. Hence, there is a need to create a diagnostic tool that could be useful in any population of patients in whom HLH is suspected [21]. In 2014, Fardet et al developed the HScore to estimate an individual’s risk of sHLH [22]. This scoring system is available free of charge on the Internet (http://saintantoine.aphp.fr/score/) and is based on 9 clinical aspects: known underlying immunodepression, maximal temperature, presence of hepatomegaly, presence of splenomegaly, hemoglobin concentration, leucocyte count, platelet count, ferritin concentration, triglyceride concentration, fibrinogen concentration, SGOT/ASAT activity, and hemophagocytosis features on bone marrow aspirate.
Treatment of HLH is challenging due to the turbulent course of the disease and should focus on 3 aspects. First, it should include treatment specific to HLH inducing factors (eg, EBV or CMV infection). Second, it should be directed at an inflammation suppression by acting on CD8+ T lymphocytes and antigen-presenting cells (APCs). Finally, in FHL, it must aim at the underlying immunodeficiency [23]. The treatment of FHL is not trivial, but is based on allogeneic stem cell transplantation in the vast majority of cases.
The present case of sHLH reported by us was characterized by an unidentified etiological factor of sHLH at the time when first symptoms occurred, lack of comorbidities, recurrent episodes of fever, and a relatively long period of nonspecific clinical course, which resulted in a late diagnosis of the disease in a young man.
Case Report
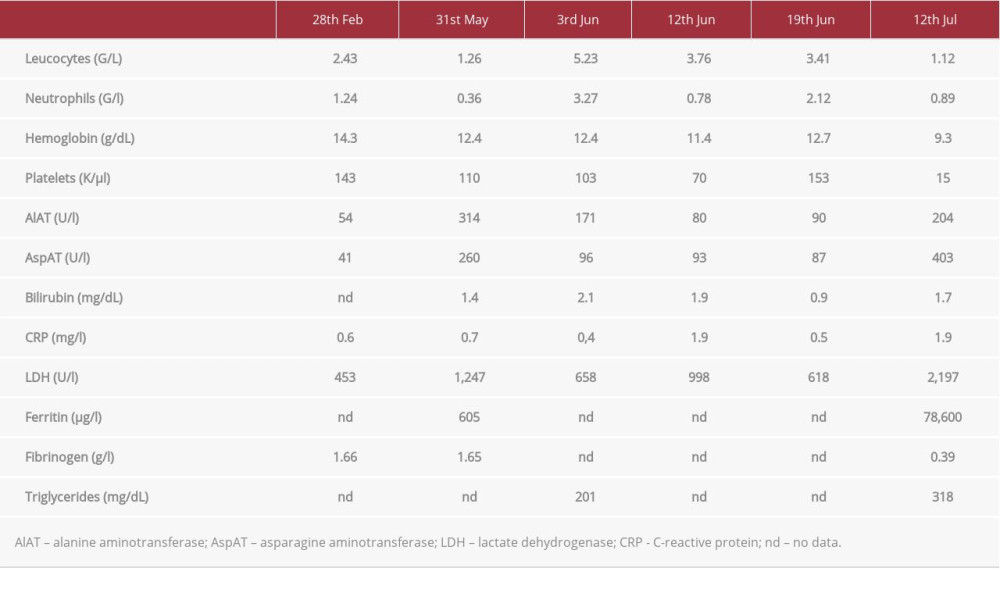
A 28-year-old man was referred to the Department of Internal Medicine in February 2019 due to hepatomegaly, splenomegaly, leukopenia, and thrombocytopenia. In the anamnesis, the patient reported having Gilbert’s syndrome, diagnosed based on clinical symptoms and laboratory markers. At the admission physical examination, a slightly enlarged spleen and liver were found as the only abnormalities. Laboratory tests revealed leukopenia with neutropenia, thrombocytopenia, slightly elevated activity of aminotransferases, and increased total bilirubin level. Basic laboratory test results at admission are presented in Table 1. Viral studies excluded human immuno-deficiency virus (HIV), hepatitis C virus (HCV), and hepatitis B virus (HBV) infection. Abdominal ultrasonography (USG) confirmed the enlargement of the liver with the length up to 15 cm and spleen up to 17 cm, but otherwise no significant abnormalities were revealed. Then, patient was referred to our Hematology Clinic to extend diagnostic procedures. Bone marrow smear evaluation and multiparameter flow cytometric analysis revealed no abnormalities. The bone marrow cytogenetic study was undiagnostic due to low cell proliferation. In March 2019, a positron emission tomography (PET-CT) examination was performed and showed no foci of increased (18) F-fluorodeoxyglucose (18 F-FDG) metabolism.
At the end of May 2019, the patient was admitted to the Department of Infectious Diseases due to fever that lasted for several days and a persistent dry cough. The physical examination again showed no abnormalities other than an enlarged liver and spleen. Urine culture revealed
Another bone marrow examination was performed, again confirming the normal cytomorphology and immunophenotype of bone marrow cells. There were no ring sideroblasts in the bone marrow smear. In addition, paroxysmal nocturnal hemoglobin-uria was also excluded by flow cytometry evaluation of glycosylphosphatidylinositol-linked proteins expression on leukocytes in peripheral blood. Cytomegalovirus (CMV), EBV, and parvovirus B19 infections were excluded by molecular tests several times at each admission to the hospital. Testing for beta-glucocerebrosidase and ceruloplasmin activity was normal, as was a chest X-ray. The patient was treated empirically with levofloxacin and filgrastim. He recovered from fever, and the leukocyte count increased (Table 1).
From June 12, 2019, the patient experienced recurrence of fever and worsening of neutropenia (Table 1). Filgrastim and prophylactic antibiotic therapy were restarted. However, again, no infection was documented either clinically or microbiologically. Since autoimmune disease was suspected, steroid therapy was initiated while awaiting the results of antibody tests. The starting dose was 30 mg prednisone per day. Clinical improvement was achieved, fever subsided, and leukocyte count increased (Table 1). The additional tests excluded the presence of antinuclear antibody (ANA), anti-neutrophil cytoplasmic antibodies (ANCA), anti-hepatic, anti-cardiolipin, and anti-beta2-microglobulin antibodies. Due to the persistent elevated activity of aminotransferases, concentration of bilirubin, and the enlargement of liver and spleen, a liver biopsy was performed at the Department of Infectious Diseases on June 30, 2019. Before the procedure, USG showed a slight increase in the size of the liver to 18 cm. The course of the procedure was uncomplicated, and no bleeding occurred. However, 1 day after the procedure, fever recurred, without a significant increase in inflammatory parameters. Empirical antibiotic therapy was started, and the doses of steroids were escalated. However, the fever did not resolve. Histopathological examination of the liver biopsy showed no clinically important abnormalities. On July 12, 2019, patient was transferred to the Department of Hematology. On admission, his general condition was average and he presented with fever. He reported severe abdominal pain in the left upper quadrant. On physical examination, the spleen and liver were significantly enlarged, palpable approximately 10 cm below the left costal margin. Abdominal USG was performed and, apart from splenomegaly, the presence of free fluid in the abdominal cavity was found. Additional laboratory testing disclosed a rapid drop in hemoglobin concentration (Table 1). The abdominal computed tomography (CT) revealed an enlargement of the liver up to 23 cm and spleen up to 23 cm, with the presence of a subcapsular rupture in the lower pole of about 13 mm (Figures 1, 2). Moreover, numerous retroperitoneal enlarged lymph nodes were visualized.
On July 13, 2019, the patient was transferred to the Surgery Department, where splenectomy was performed and enlarged lymph nodes were collected for histopathological examination.
Then, the patient was transferred to the Intensive Care Unit (ICU), where he required numerous transfusions of erythrocytes and platelets concentrates, fresh frozen plasma (FFP), and recombinant factor VIIa (rVIIa). On July 14, 2019, the presence of a hematoma was found in the spleen extraction site, and another laparotomy was performed. Then, the patient was further treated in the ICU. The aspiration biopsy done at that time showed numerous hemophagocytes (Figure 3). There was also a significant increase in ferritinemia to over 120 000 μg/l. The patient met 5 of 8 HLH 04 criteria for diagnosis of hemophagocytic syndrome, including 2 clinical (fever and splenomegaly) and 4 laboratory (cytopenia, hyperferritinemia, hypertriglyceridemia, and hypofibrinogenemia) criteria. Due to the inability to assess NK lymphocyte function and sCD25 concentration, we were not able to assess all HLH04 criteria. Therefore, the HScore evaluation turned out to be a more useful tool, and the final HScore was 253.
Treatment was instantly initiated with dexamethasone (DEX) 10 mg/m2 daily and etoposide 100 mg/m2 (Vepesid, Vep) in a dose adjusted to estimated glomerular filtration rate (eGFR), and cyclosporine A (CSA) in a dose adjusted to the blood concentration of the drug. In total, 2 doses of Vep were given within 4 days. Despite intensive treatment, the patient’s condition deteriorated rapidly. He developed multiple-organ failure and died on July 22, 2019.
Finally, the histopathological assessment of the retroperitoneal lymph nodes revealed peripheral T-cell lymphoma not otherwise specified (PTCL-NOS). The result of histopathological lymph nodes examination was obtained after the patient’s death.
Discussion
The current guidelines with revised diagnostic criteria for sHLH in adults without rheumatoid disease were introduced in 2004 [21]. However, using the HLH-2004 criteria for adult patients with M-HLH remains controversial. In such cases, it seems more useful to use the HScore. The patient described in this report achieved an HScore of 254 points and probability of having HLH about 99%. Although the HScore in patients with M-HLH is usually lower than in patients with other sub-types of HLH, in our patient it reached a very high value [24]. Of note, the final diagnosis of PTCL-NOS was established after the patient’s death, based on histopathological evaluation of the retroperitoneal lymph nodes. Since the retroperitoneal lymph nodes were the only enlarged ones, this delayed the diagnosis of lymphoma.
An interesting approach to the diagnosis of HLH was also presented by Smits et al [25]. The authors divided the HLH 04 criteria into 2 major (splenomegaly, presence of hemophagocytosis in tissue samples) and 3 minor (presence of >1 cytopenia, ferritin concentration >1000 µg/l, and a decrease in fibrinogen concentration with increased triglycerides). Meeting 1 major and 1 minor criterion gives a chance of early diagnosis of HLH, with a sensitivity of 79% and specificity of 97%. Unfortunately, at an early stage, our patient did not meet all the minor criteria (fibrinogen >1 g/l and ferritin <1000 ug/l), thus this approach would not allow for a correct diagnosis.
Due to diagnostic difficulties, works are in progress to identify new diagnostic markers for HLH. Ren et al performed studies using enzyme-linked immunosorbent assay (ELISA) on HLH patients with NK/T cell lymphoma [26]. They concluded that platelet factor 4 (PF4) and angiopoietin-4 (ANG-4) elevated in the serum and downregulated platelet-derived growth factors (PDGFs) may be useful biomarkers for early detection of NK/TLAHS. LAHS is a highly lethal immunological disorder in which the poor prognosis is partly due to a late diagnosis. Frequent lack of lymphadenopathy or tumor mass from which the biopsy could be performed makes the diagnosis difficult. The pathogenesis of LAHS includes inflammation, loss of immune balance due to infection or chemotherapy, and permanent stimulation of antigen by neoplastic cells, resulting in secretion of large amounts of cytokines. Lack of effective treatment if the underlying cause of HLH is not diagnosed can lead to a rapid and fatal course. Patients with LAHS have a shorter overall survival (OS) and a higher early death rate compared to patients with the other subtypes of HLH. Early diagnosis of lymphoma improves the prognosis [27].
In case of PTCL-NOS, the clinical manifestations can vary widely. A case of PTCL of the colon associated with HLH has already been described, with a clinical course like Crohn’s disease [28]. The patient, similarly to the present patient, had abdominal pain for 1 month and recurrent fever. Moreover, he had no superficial lymphadenopathy, and an infectious etiology of symptoms, such as CMV, EBV, and tuberculosis, was suspected, but was finally excluded. Tests also revealed leukopenia, severe anemia, and thrombocytopenia. CT examination revealed hepatosplenomegaly, but without enlarged lymph nodes, and biopsy of the bone marrow confirmed HLH. Due to intestinal perforation near the splenic flexure with peritonitis, the patient was operated on, and a histopathological examination revealed PTCL. The patient died due to sepsis. Liu et al described a group of patients who were diagnosed with NK/T cell LAHS and presented with fever (96.4%), splenomegaly (81.5%), hyperferritinemia (91.7%), hypertriglyceridemia (48%), severe anemia (46.4%), and hypofibrinogenemia (45%). In this study, the mortality rate was remarkably high (96.4%), with low fibrinogen level as an adverse prognostic factor [29]. The same was observed for high LDH level and splenomegaly. In addition, Jin et al conducted a study in which they analyzed the prognostic factors in T/NK-LAHS disease [30]. They found that EBV-positive patients had poor prognosis, as did those who do not receive allogeneic hematopoietic stem cell transplantation (allo-HSCT), and did not achieve early overall response after initial induction therapy. These patients died within 1 month. The authors reported that NK/T-LAHS is a high-mortality disease with poor prognosis. Bigenwald et al suggested that LAHS patients have poorer prognosis and die earlier if they are not treated with etoposide [31].
The diagnosis of HLH is made difficult by its clinical course being similar to that of sepsis, systemic inflammatory response syndrome, multiple-organ dysfunction syndrome (MODS), Kawasaki’s disease, and severe infection [32]. Bicytopenia, fever, hypertriglyceridemia, hypofibrinogenemia, and hyperferritinemia are often associated with sepsis, SIRS, and MODS. Hemophagocytosis particularly occurs in patients with SIRS and in children and adolescents with MODS. The diagnosis of HLH is usually based on nonspecific clinical features. Moreover, diagnostic abnormalities in HLH may appear with a delay, which can make differential diagnosis difficult. Patients with sepsis, SIRS, or MODS may meet the current diagnostic criteria for HLH and therefore may be subject to inappropriate treatment. To increase the number of appropriate and early diagnoses, scientists are looking for pathognomonic criteria for HLH. A prospective study of 756 children in a hematological oncology department was conducted to create a panel of HLH-specific cytokines, showing that the Th1/Th2 cytokine pattern was different between HLH and infections. Moreover, high diagnostic accuracy for HLH among children with fever showed high levels of IFN-gamma and IL-10 with moderately elevated IL-6 and proved useful to differentiate HLH from infection, in addition to ferritin, a well-known laboratory parameter helpful in diagnosing HLH [33].
HLH should be suspected in patients with high continuous fever of unknown origin and enlarged internal organs. Early diagnosis is crucial for implementing appropriate and effective treatment. Unfortunately, there are no pathognomonic tests for HLH [16]. Unfortunately, hemophagocytosis is not an essential diagnostic element of HLH, since this condition may be also observed in infections [34]. Hyperferritinemia often accompanies rapidly progressing interstitial lung disease (ILD) with polymyositis (PM) or dermatomyositis (DM) [35]. It can also occur in other autoimmune diseases such as Still’s disease, rheumatoid arthritis, and SLE. Moreover, it occurs in patients with atherosclerosis, Parkinson’s disease, Alzheimer’s, restless leg syndrome, malignant tumors, and Gaucher’s disease [36,37]. The authors of a study conducted in the USA based on a cohort of 73 patients with HLH indicate a triad of symptoms that should suggest a diagnosis of HLH; it consists of cytopenias, fever, and hyperferritinemia [16,38].
HLH treatment is based on a combination of chemotherapeutic and immunosuppressive drugs to reduce proliferation and activation of immune system cells and inhibit cytokine storm. The systemic treatment includes dexamethasone (DEX), etoposide, and cyclosporine A (CSA) [39].
The treatment of a patient with diagnosed HLH should be multidisciplinary, and therapy specific to the underlying disease alone is usually not sufficient. The HLH-2004 protocol (NCT00426101) compared to the HLH-94 protocol contains CSA in induction from the first day onwards [21,40]. The HLH-94 protocol, which includes the administration of etoposide and DEX together with the use of CSA for maintenance treatment, should be applied in patients with severe HLH with an unknown trigger. The use of etoposide stops irreversible organ destruction by macrophages and cytotoxic CD8+ lymphocytes. The efficacy of etoposide and DEX in combination with CSA is confirmed by clinical studies, but only in patients aged <18 years. There are currently 81 clinical trials registered in the ClinicalTrials.gov database on HLH, 66 of which are conducted in adults (18–64 years old). Some of them include tocilizumab (NCT02007239), etoposide and DEX (NCT03117010), or splenectomy (NCT02862054). Performing a splenectomy is one of the methods of supportive treatment, which results in a median survival time of 22 months [40]. However, post-splenectomy complications in HLH patients include massive bleeding and infections (eg, pulmonary). One of the reported patients experienced oozing of blood from the wound, and massive FFP, platelets, and similar transfusions were not effective [41]. This may suggest that the patient also experienced post-splenectomy complications, which in turn referred the diagnosis to HLH.
Authors from Japan published a retrospective single-center study of 34 patients with HLH diagnosed between 2001 and 2014 according to HLH-2004 guidelines. After a median follow-up of 1.9 months, 11 of the 34 patients died. Multi-organ failure, respiratory failure, septic shock, malignant tumors, liver failure, and cerebral hemorrhage were among the causes of death. It turned out that age at the diagnosis was the strongest prognostic factor regardless of HLH etiology, with younger patients who survived [42]. On the other hand, Chang et al concluded that patients with LAHS have the worst prognosis, and there is no difference in survival between patients with B cell and T/NK cell LAHS [43]. It should also be noted that until the treatment protocols were introduced, the 1-year survival was close to 0%, and following the introduction of HLH-94 protocol treatment survival in sHLH it increased to 55%. The possibility of allo-HSCT with reduced intensity conditioning (RIC) is also promising [44,45].
Currently, the therapeutic approach is changing, immunosuppression is being abandoned, and treatment is more often being used to targeted cytokines, selected cells, and signaling pathway points, which have their place in HLH pathophysiology. An alternative treatment that may be offered to patients not responding to conventional treatment are biological drugs. In those cases, treatment should be “tailored” to the specific clinical situation [46]. Tocilizumab (IL- 6 inhibitor) may be successfully used in patients with COVID-19 [47]. Another interesting example is the effective treatment of HLH with central nervous system (CNS) involvement with the use of the IL-1b inhibitor [48]. DEX in combination with human monoclonal anti-IFN-gamma antibody also seems to be promising as a second-line treatment in patients with primary HLH. Other treatment options include anti-thymocyte globulins, alemtuzumab, anti-CD52 antibody, daclizumab, and anti-TNF alpha. Personalization of the approach and targeted therapies are likely to improve patient prognosis [49].
Conclusions
In conclusion, HLH still remains difficult to diagnose and treat, which is reflected in the high mortality rate of 52-75% reported in the literature. The decision to carry out genetic tests as part of diagnostics should be considered individually if no identifiable causes of the disease have been established [38]. The prognosis is worse for males and may be improved by rapid implementation of treatment after a diagnosis based on a bone marrow biopsy or an organ showing hemophagocytosis [34,50,51].
Despite availability of more modern methods of treatment, as well as new prognostic and predictive markers for HLH, many cases are still diagnosed too late. Unfortunately, the present patient was also undiagnosed for several months due to un-specific symptoms and signs.
Prompt and accurate diagnosis of HLH is critical to the implementation of appropriate treatment. Delay in treatment can lead to organ complications, a severe course of the disease, and patient death.
Figures
References:
1.. Bodley Scott R, Robb-Smith AHT, Histiocytic medullary reticulosis: Lancet, 1939; 234(6047); 194-98
2.. Farquhar JW, Claireaux AE, Familial haemophagocytic reticulosis: Arch Dis Child, 1952; 27(136); 519-25
3.. Schulert GS, Grom AA, Macrophage activation syndrome and cytokine-directed therapies: Best Pract Res Clin Rheumatol, 2014; 28(2); 277-92
4.. George MR, Hemophagocytic lymphohistiocytosis: Review of etiologies and management: J Blood Med, 2014; 5; 69-86
5.. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Perforin gene defects in familial hemophagocytic lymphohistiocytosis: Science, 1999; 286(5446); 1957-59
6.. Esteban YM, de Jong JLO, Tesher MS, An overview of hemophagocytic lymphohistiocytosis: Pediatr Ann, 2017; 46(8); e309-13
7.. Wang Y, Wang Z, Zhang J, Genetic features of late onset primary hemophagocytic lymphohistiocytosis in adolescence or adulthood: PLoS One, 2014; 9(9); e0107386
8.. Marsh RA, Haddad E, How I treat primary haemophagocytic lymphohistiocytosis: Br J Haematol, 2018; 182(2); 185-99
9.. Zain JM, Aggressive T-cell lymphomas: 2019 updates on diagnosis, risk stratification, and management: Am J Hematol, 2019; 94(8); 929-46
10.. Swerdlow SH, Campo E, Pileri SA, The 2016 revision of the World Health Organization classi fi cation of lymphoid neoplasms: Blood, 2016; 127(20); 2375-91
11.. Lehmberg K, Nichols KE, Henter J-I, Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies: Haematologica, 2015; 100(8); 997-1004
12.. Ishii E, Ohga S, Imashuku S, Nationwide survey of hemophagocytic lymphohistiocytosis in Japan: Int J Hematol, 2007; 86(1); 58-65
13.. Machaczka M, Vaktnäs J, Klimkowska M, Hägglund H, Malignancy-associated hemophagocytic lymphohistiocytosis in adults: A retrospective population-based analysis from a single center: Leuk Lymphoma, 2011; 52(4); 613-19
14.. Mehta P, McAuley DF, Brown M, COVID-19: Consider cytokine storm syndromes and immunosuppression: Lancet, 2020; 395(10229); 1033-34
15.. Eroglu A, Kartal S, Saral OB, Helmet mask and tocilizumab for a patient with hemophagocytic lymphohistiocytosis syndrome and COVID-19: A case report: Brazilian J Anesthesiol, 2021; 71(1); 79-83
16.. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Adult haemophagocytic syndrome: Lancet, 2014; 383(9927); 1503-16
17.. Crayne CB, Albeituni S, Nichols KE, Cron RQ, The immunology of macrophage activation syndrome: Front Immunol, 2019; 10; 119
18.. Henderson LA, Cron RQ, Macrophage activation syndrome and secondary hemophagocytic lymphohistiocytosis in childhood inflammatory disorders: Diagnosis and management: Paediatr Drugs, 2020; 22(1); 29-44
19.. Boom V, Anton J, Lahdenne P, Evidence-based diagnosis and treatment of macrophage activation syndrome in systemic juvenile idiopathic arthritis: Pediatr Rheumatol Online J, 2015; 13; 55
20.. Hayden A, Park S, Giustini D, Lee AYY, Chen LYC, Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic scoping review: Blood Rev, 2016; 30(6); 411-20
21.. Henter J-I, Horne A, Aricó M, HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis: Pediatr Blood Cancer, 2007; 48(2); 124-31
22.. Fardet L, Galicier L, Lambotte O, Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome: Arthritis Rheumatol, 2014; 66(9); 2613-20
23.. Henter J-I, Biology and treatment of familial hemophagocytic lymphohistiocytosis: Importance of perforin in lymphocyte-mediated cytotoxicity and triggering of apoptosis: Med Pediatr Oncol, 2002; 38(5); 305-9
24.. Debaugnies F, Mahadeb B, Ferster A, Performances of the H-score for diagnosis of hemophagocytic lymphohistiocytosis in adult and pediatric patients: Am J Clin Pathol, 2016; 145(6); 862-70
25.. Smits BM, van Montfrans J, Merrill SA, A minimal parameter set facilitating early decision-making in the diagnosis of hemophagocytic lymphohistiocytosis: J Clin Immunol, 2021; 41(6); 1219-28
26.. Ren Q, Chan K-W, Huang H, Platelet-derived alpha-granules are associated with inflammation in patients with NK/T-cell lymphoma-associated hemophagocytic syndrome: Cytokine, 2020; 126; 154878
27.. Xie M, Li L, Zhu L, An effective diagnostic index for lymphoma-associated hemophagocytic syndrome: QJM, 2018; 111(8); 541-47
28.. Tsai A-S, Ko C-W, Yeh H-Z, Peripheral T-cell lymphoma of the colon associated with hemophagocytic lymphohistiocytosis: J Chin Med Assoc, 2013; 76(3); 169-72
29.. Liu Y-Z, Bi L-Q, Chang G-L, Clinical characteristics of extranodal NK/ T-cell lymphoma-associated hemophagocytic lymphohistiocytosis: Cancer Manag Res, 2019; 11; 997-1002
30.. Jin Z, Wang Y, Wang J, Multivariate analysis of prognosis for patients with natural killer/T cell lymphoma-associated hemophagocytic lymphohistiocytosis: Hematology, 2018; 23(4); 228-34
31.. Bigenwald C, Fardet L, Coppo P, A comprehensive analysis of Lymphoma-associated haemophagocytic syndrome in a large French multicentre cohort detects some clues to improve prognosis: Br J Haematol, 2018; 183(1); 68-75
32.. Xu X, Tang Y, Song H, Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children: J Pediatr, 2012; 160; 984-90.e1
33.. Tang Y-M, Xu X-J, Advances in hemophagocytic lymphohistiocytosis: Pathogenesis, early diagnosis/differential diagnosis, and treatment: Scientific World Journal, 2011; 11; 697-708
34.. Florena AM, Iannitto E, Quintini G, Franco V, Bone marrow biopsy in hemophagocytic syndrome: Virchows Arch, 2002; 441(4); 335-44
35.. Kawasumi H, Gono T, Kawaguchi Y, IL-6, IL-8, and IL-10 are associated with hyperferritinemia in rapidly progressive interstitial lung disease with polymyositis/dermatomyositis: Biomed Res Int, 2014; 2014; 815245
36.. Zandman-Goddard G, Shoenfeld Y, Hyperferritinemia in autoimmunity: Isr Med Assoc J, 2008; 10(1); 83-84
37.. Stein P, Yu H, Jain D, Mistry PK, Hyperferritinemia and iron overload in type 1 Gaucher disease: Am J Hematol, 2010; 85(7); 472-76
38.. Otrock ZK, Eby CS, Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis: Am J Hematol, 2015; 90(3); 220-24
39.. Bergsten E, Horne A, Aricó M, Confirmed efficacy of etoposide and dexamethasone in HLH treatment: Long-term results of the cooperative HLH-2004 study: Blood, 2017; 130(25); 2728-38
40.. Henter J-I, Samuelsson-Horne A, Aricò M, Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation: Blood, 2002; 100(7); 2367-73
41.. Jing-Shi W, Yi-Ni W, Splenectomy as a treatment for adults with relapsed hemophagocytic lymphohistiocytosis of unknown cause: Ann Hematol, 2015; 94(5); 753-60
42.. Oto M, Yoshitsugu K, Uneda S, Prognostic factors and outcomes of adult-onset hemophagocytic lymphohistiocytosis: A retrospective analysis of 34 cases: Hematol Rep, 2015; 7(2); 5841
43.. Chang Y, Cui M, Fu X, Lymphoma associated hemophagocytic syndrome: A single-center retrospective study: Oncol Lett, 2018; 16(1); 1275-84
44.. Malinowska I, Machaczka M, Popko K, Hemophagocytic syndrome in children and adults: Arch Immunol Ther Exp (Warsz), 2014; 62(5); 385-94
45.. Messina C, Zecca M, Fagioli F, Outcomes of children with hemophagocytic lymphohistiocytosis given allogeneic hematopoietic stem cell transplantation in Italy: Biol Blood Marrow Transplant, 2018; 24(6); 1223-31
46.. Rosée P La, Horne AC, Hines M, Recommendations for the man -agement of hemophagocytic lymphohistiocytosis in adults: Blood, 2019; 133(23); 2465-77
47.. Tholin B, Hauge MT, Aukrust P, Hemophagocytic lymphohistiocytosis in a patient with COVID-19 treated with tocilizumab: A case report: J Med Case Rep, 2020; 14(1); 1-5
48.. Hiraldo JDG, Domínguez-Mayoral A, García-Gómez FJ, Central nervous system involvement in adult-onset relapsing hemophagocytic lymphohistiocytosis responsive to maintenance treatment with anakinra: J Neuroimmunol, 2021; 355; 577552
49.. Brisse E, Matthys P, Wouters CH, Understanding the spectrum of haemophagocytic lymphohistiocytosis: Update on diagnostic challenges and therapeutic options: Br J Haematol, 2016; 174(2); 175-87
50.. Li J, Wang Q, Zheng W, Hemophagocytic lymphohistiocytosis: Clinical analysis of 103 adult patients: Medicine (Baltimore), 2014; 93(2); 100-5
51.. Parikh SA, Kapoor P, Letendre L, Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis: Mayo Clin Proc, 2014; 89(4); 484-92
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133