08 October 2022: Articles 
Lupus Anticoagulant-Hypoprothrombinemia Syndrome and Pseudotumor Cerebri as an Initial Presentation of Systemic Lupus Erythematosus in a 16-Year-Old Male Patient: A Case Report and Literature Review
Challenging differential diagnosis, Unusual or unexpected effect of treatment, Rare disease, Rare coexistence of disease or pathology
Mohamed H. Omer1DEF, Hind SalamaDOI: 10.12659/AJCR.938051
Am J Case Rep 2022; 23:e938051






Abstract
BACKGROUND: Lupus anticoagulant-hypoprothrombinemia syndrome (LAHPS) is an exceptionally rare disease caused by prothrombin antibodies, resulting in reduced factor II levels. This disease can present with significant bleeding and is usually associated with autoimmune disorders, particularly systemic lupus erythematosus (SLE). There are currently no guidelines for the treatment of LAHPS, and corticosteroids remain the criterion standard therapy. Pseudotumor cerebri is a disease that involves an idiopathic rise in intracranial pressure in association with papilledema. The coexistence of pseudotumor cerebri with SLE is rare, with an overall incidence of 0.7%.
CASE REPORT: A 16-year-old male initially presented to our hospital with nausea, headaches, and decreased visual acuity. He was diagnosed with pseudotumor cerebri based on the findings of papilledema and a raised opening pressure on lumbar puncture. Three months later, he presented with macroscopic hematuria and persistent epistaxis. Further investigation revealed a prolonged activated partial thromboplastin time and prothrombin time, along with positive LA and reduced Factor II levels, resulting in a diagnosis of LAHPS. The patient received a dose of 1 mg/kg/day of prednisolone along with hydroxychloroquine, and he had a complete recovery with cessation of bleeding and normalization of laboratory parameters.
CONCLUSIONS: We are reporting a case of pseudotumor cerebri with a further presentation of LAHPS in a patient found to have SLE. As both associations are rare in the presence of SLE, it is vital to recognize them early to initiate adequate management and intervention to avoid life-threatening complications.
Keywords: Acquired Hypoprothrombinemia, Hemostasis, Lupus Erythematosus, Systemic, Pseudotumor Cerebri, Thrombosis, Adolescent, Adrenal Cortex Hormones, Antiphospholipid Syndrome, Hemorrhage, Humans, hydroxychloroquine, Hypoprothrombinemias, Lupus Coagulation Inhibitor, Male, papilledema, Prednisolone, Prothrombin
Background
Prothrombin is a vitamin K-dependent coagulation cofactor. Once activated, prothrombin is proteolytically cleaved by factor Xa to form thrombin [1]. Thrombin induces platelet aggregation and results in the activation of different mediators required in the clotting cascade. Prothrombin deficiency can be due to either an inherited or acquired etiology. An acquired prothrombin deficiency can result from a multitude of factors, including end-stage liver disease, malignancy, viral infection, vitamin K deficiency, and autoimmune conditions [2]. However, in many of these scenarios, a deficiency in other coagulation factors is noted.
Systemic lupus erythematosus (SLE), an autoimmune inflammatory disorder, tends to be associated with antiphospholipid syndrome (APLS), which is a systemic inflammatory disease characterized by the presence of lupus anticoagulants and the incidence of thrombotic events [3]. Lupus anticoagulant is an antiphospholipid antibody that tends to inhibit phospholipid-dependent clotting without impacting the activity of other clotting factors, thereby resulting in thrombotic events [4]. In rare instances, the presence of an acquired prothrombin deficiency may be observed in patients with lupus anticoagulant (LA) [5]. This association tends to be primarily seen in patients with SLE and is referred to as lupus anticoagulanthypoprothrombinemia syndrome (LAHPS) [6]. Unlike patients with APLS, patients with LAHPS tend to present with frequent bleeding episodes ranging from mild mucocutaneous bleeds to life-threatening intracranial hemorrhages [7].
Pseudotumor cerebri (also known as idiopathic intracranial hypertension) is a condition characterized by a rise in intracranial pressure accompanied by papilledema in the absence of a space-occupying lesion and abnormal cerebrospinal fluid components [8]. A few cases of patients with SLE presenting with pseudotumor cerebri have been described in the literature, with the association having a reported incidence of 0.7% [9].
Herein we report a case of a 16-year-old male patient with pseudotumor cerebri and LAHPS with a background of systemic lupus erythematosus.
Case Report
A 16-year-old Saudi-Arabian male with no prior past medical history presented to the Emergency Department with a one-week history of severe bilateral headaches associated with nausea and a decrease in visual acuity. Upon examination, the patient had a body mass index of 30 kg/m2. In addition, neurological examination revealed an absence of focal neurological signs. Ophthalmic examination revealed corrected visual acuity of 20/30 in the left eye and 20/25 in the right eye. Fundoscopic examination revealed bilateral optic disc swelling consistent with papilledema. Computed tomography (CT) scans of the brain along with a CT venogram were normal. A diagnostic lumbar puncture was performed, and the opening pressure was stated as 40 cmH2O (normal range 6–25 cm-H2O). Analysis of the cerebrospinal fluid for protein, glucose, cell count, along with viral, bacterial, and fungal cultures did not yield any results. Laboratory parameters including a complete blood count, coagulation studies, and liver function tests were reported as normal. Therefore, in line with the diagnostic criteria for pseudotumor cerebri due to the presence of papilledema and a raised opening pressure on lumbar puncture in the absence of signs of space-occupying lesions and focal neurological signs, the patient was diagnosed with pseudo-tumor cerebri [8].
Treatment was initiated with oral acetazolamide at a dose of 250 mg per day. The patient followed up with the Neurology Department after initiation of treatment and experienced a rapid course of recovery with improvement in symptoms and visual acuity along with significant improvement in the degree of papilledema.
Two months after his initial admission, the patient presented to the Emergency Department with a 1-month history of recurrent epistaxis and macroscopic hematuria. There was no history of fevers, night sweats, ecchymosis, or bleeding from any other sites. However, the patient recounted a 1-month history of arthralgia and generalized fatigue. A physical examination revealed bilateral cervical lymphadenopathy with no other physical signs. Initial investigations revealed a hemoglobin of 9.5 g/dL (reference range 13.20–16.60), a white cell count of 5.5×109/L (reference range 4.50–11.00), and a platelet count of 184×109/L (reference range 150–400). Moreover, the patient had an elevated erythrocyte sedimentation rate of 55 mm/h (reference range <15). Liver function tests, blood urea and nitrogen, and creatinine tests were within normal limits. A computed tomography scan of the abdomen and pelvis was undertaken to investigate the macroscopic hematuria, which was reported as normal. Furthermore, a lymph node biopsy of the enlarged cervical lymph nodes revealed reactive lymphoid tissue with no evidence of malignancy.
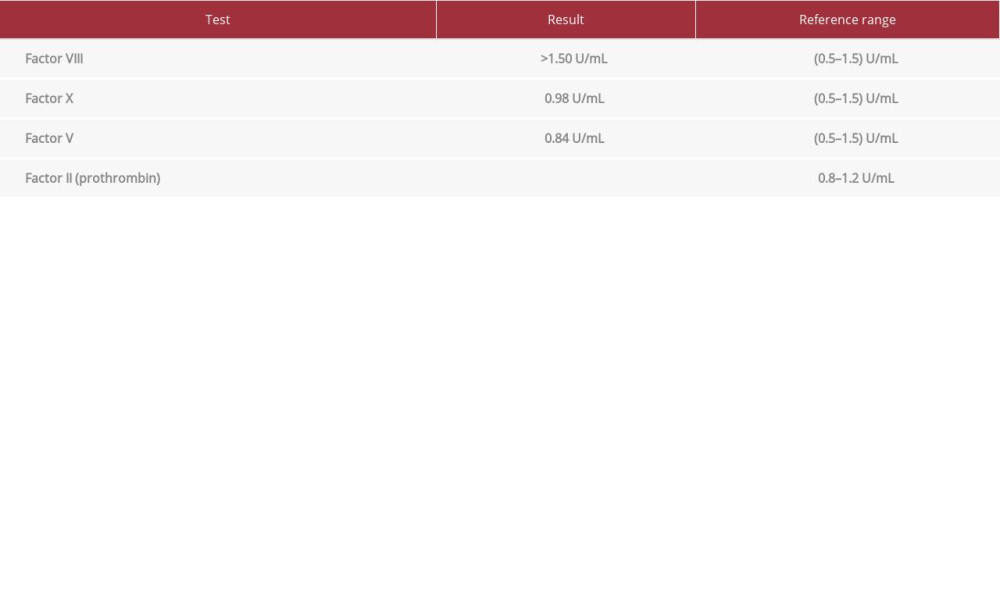
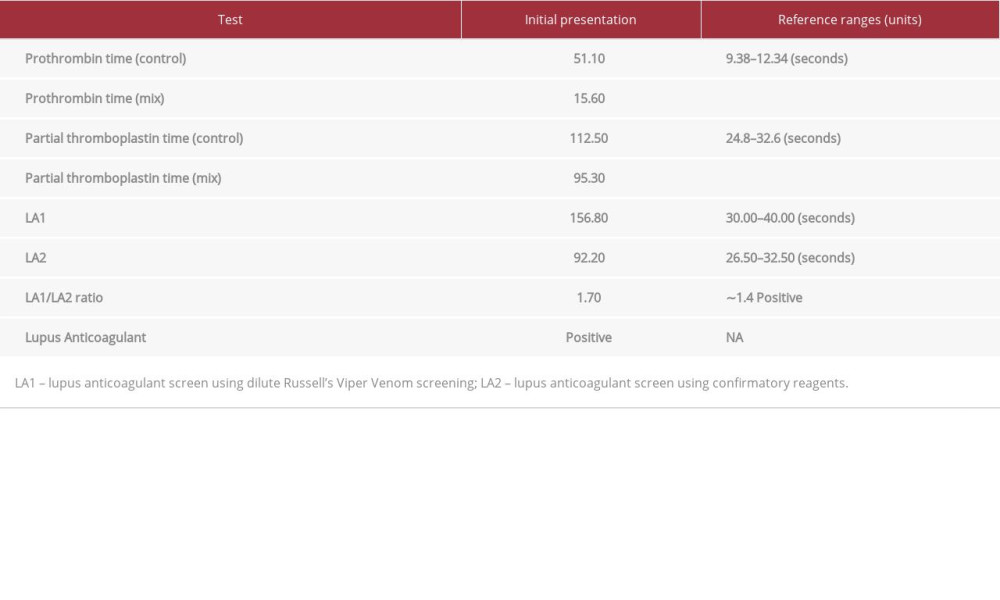
Coagulation studies illustrated a markedly prolonged partial thromboplastin time (PTT) of 112.5 seconds (reference range 24.80–32.60) and a prolonged prothrombin time (PT) of 51.1 s (reference range 9.38–12.34). A factor assay test was conducted to determine the levels of coagulation factors, revealing a severe isolated deficiency in factor 2 levels with a level of <0.06 U/mL (reference range 0.8–1.2). Table 1 summarizes the findings of the factor assay showing an isolated factor II deficiency. The prolongation of PTT and PT along with the low levels of factor 2 may result from a coagulation factor deficiency or the presence of an inhibitor. To differentiate between the aforementioned causes of the coagulation anomalies, we performed a mixing study which involves mixing the patient’s plasma with a pooled normal plasma sample acting as a control at a 1: 1 ratio. After the mixing study, the PTT and PT levels failed to correct, suggesting that the low levels of factor 2 were due to the presence of an inhibitor. To identify the inhibitor, we performed a test for lupus anticoagulant, an autoantibody which often interferes with phospholipid-dependent tests such as the PTT [11]. Using the dilute Russel viper venom test followed by a confirmatory reagent test, the presence of lupus anticoagulant was confirmed. Table 2 summarizes the findings of the 1: 1 mixing study and the lupus anticoagulant detection tests.
Moreover, additional investigations revealed a positive antinu-clear antibody test at 176.85 U (>60 U is positive) and a borderline anti-double stranded DNA antibody (anti-dsDNA) level of 77.7 IU/mL (<70 IU/mL is negative). The patient also had strongly positive anti-smith antibodies (anti-SM) at 152.80 U (>80 U is strongly positive) and positive antinuclear ribonucleoprotein (anti-RNP) antibodies at 137.25 (>80 U is strongly positive). In addition, there was a low level of complement 3 (C3), at 0.19 g/L (reference range 0.7–1.52), along with an undetectable level of complement 4 (C4), at a level of <0.02 g/L (reference range 0.16–0.38).
The patient’s clinical criteria and immunological criteria were sufficient to establish a diagnosis of SLE in accordance with the 2019 EULAR/ACR classification criteria for SLE [11]. Moreover, due to the history of bleeding, low levels of factor 2, the positivity of lupus anticoagulant antibodies, and the mixing studies confirming the presence of an inhibitor, the patient was subsequently diagnosed with LAHPS secondary to SLE.
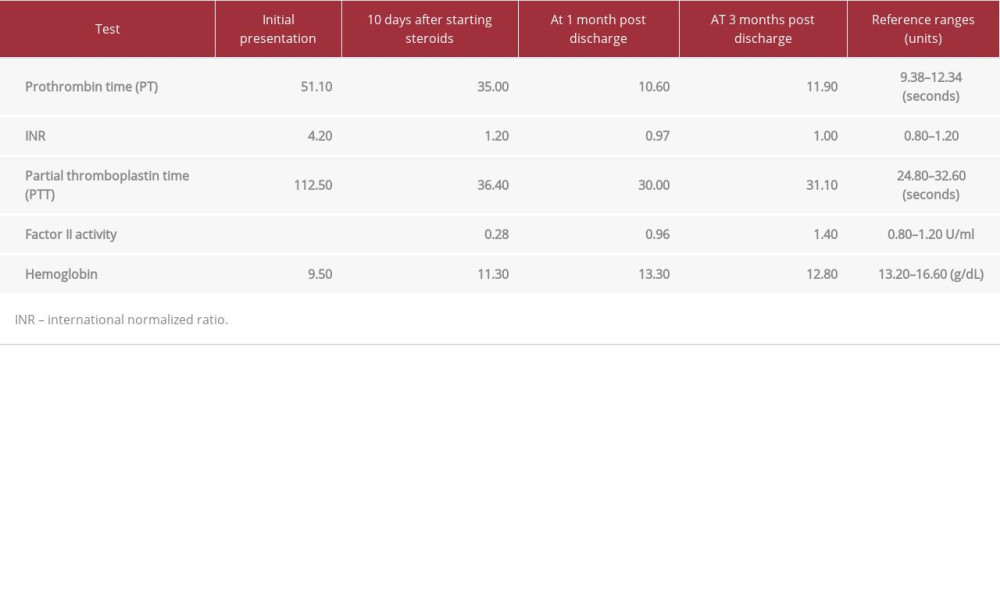
The patient received 80 mg of prednisolone per day at a 1 mg/ kg dose. After 10 days of corticosteroid therapy, the patient’s PTT and PT normalized. He began receiving hydroxychloroquine therapy at a dose of 400 mg daily and corticosteroid therapy was slowly weaned off. The patient was discharged after 10 days due to the cessation of bleeding and normalization of laboratory values. He was followed up for a period of 3 months after his discharge. The coagulation studies and hemoglobin levels remained stable for the entire period of follow-up. Table 3 summarizes the changes in the patient’s laboratory values during admission, at discharge, after 1 month of follow-up, and at 3 months of follow-up. The patient remains compliant with his treatment of 400 mg of hydroxychloroquine daily and has reported cessation of all bleeding along with significant improvements in his fatigue and arthralgia. He remains on acetazolamide therapy for his prior diagnosis of pseudotumor cerebri.
Discussion
Systemic lupus erythematosus is an autoimmune inflammatory condition that tends to be associated with antiphospholipid syndrome, resulting in several thrombotic complications. The presence of the antiphospholipid immunoglobulin lupus anticoagulant increases clotting times. However, often paradoxically, LA results in a prothrombotic state, placing the patient with SLE at risk for thrombosis [12]. In rare instances, patients with SLE present with persistent bleeding, which may be attributed to either immune-mediated thrombocytopenia as a result of infection or, in exceedingly rare scenarios, an acquired clotting factor deficiency. An acquired prothrombin deficiency associated with LA is termed lupus anticoagulant-hypoprothrombinemia syndrome [7].
LAHPS has been reported in just over 100 cases in the literature. The first case was reported in 1960 in an 11-year-old girl who presented with persistent bleeding and was found to have APLS and a low prothrombin level [13]. LAHPS tends to be seen primarily in young female patients. A review of 74 cases of LAHPS by Karin et al demonstrated that 55% of patients were under the age of 15, and 58% of the patients were female [14]. Furthermore, a more recent case series of 89 patients by Mulliez et al demonstrated that the median age of diagnosis in these patients was 12 years, with a female-to-male ratio of 1.5 to 1 [15]. In our case report, the patient did not precisely fit the pattern of patients reported in the literature, as he was male and slightly older than the mean age of diagnosis.
The pathophysiology of LAHPS is still poorly understood, with the primary proposed mechanism being the presence of auto-antibodies that bind prothrombin without neutralizing its coagulant activity, resulting in the rapid clearance of the prothrombin antibody-antigen complex, leading to escalated clearance of prothrombin from the blood [6]. However, LAHPS has also been described in patients who are negative for prothrombin antibodies, thereby highlighting the complexity and ambiguity of the pathophysiology of LAHPS [7].
The presentations of LAHPS can vary significantly, from minor mucocutaneous bleeding to life-threatening bleeding at critical sites. Mulliez et al illustrated that the most common manifestations of LAHPS among 89 patients were ecchymosis and epistaxis, seen in 44% and 35% of patients, respectively. This was followed by macroscopic hematuria and gynecological bleeding, which were seen in 15% and 14% of patients. Life-threatening bleeds such as intracranial hemorrhages and persistent surgical bleeding were rare and only observed in 6% of patients [15]. Furthermore, in another review, thrombosis was reported in 13% of cases after the initial bleeding presentation of LAHPS, thereby highlighting the need for closer follow-up of LAHPS patients [14]. Our patient presented with persistent bilateral epistaxis and frank hematuria, a reasonably common presentation amongst LAHPS patients.
LAHPS tends to be most commonly associated in the literature with autoimmune conditions, with a literature review reporting that 55% of cases were associated with an autoimmune disorder, most notably SLE in around 40% of patients [14,15]. Furthermore, 33% of patients in the same review presented with a background of infection, while 5% of patients were reported to have a background of malignancy [14,15]. Moreover, several laboratory parameters tend to be deranged during the investigations for LAHPS. Mulliez et al found that the most common coagulation defect in LAHPS tends to be a prolonged PTT and PT seen in all cases. Additionally, LA was positive in 99% of patients, whereas anticardiolipin antibodies were positive in 75%. The median value of prothrombin level in patients with LAHPS was 12% [15]. Our patient presented with LAHPS and was subsequently diagnosed with SLE. He also demonstrated significantly elevated PT and PTT times, with a prothrombin level of 0.06 U/mL. This is in keeping with patterns reported in the literature. Interestingly, our patient also had strong positive anti-Smith muscle antibodies and antinuclear ribonucleoprotein antibody titers. In a case control study of 63 patients, anti-Smith and anti-RNP antibodies were significantly associated with thrombosis amongst patients with SLE (P=0.001) [16]. Furthermore, the patient had negative IgG anticardiolipin antibodies. This was a surprising finding as approximately 75% of patients with LAHPS were found to have strong positive titers of IgG anticardiolipin antibodies [15]. In a systematic review of patients with APLS, lupus anticoagulants were deemed to be stronger risk factors of thrombosis than anticardiolipin antibodies [17]. The presence of anti-RNP antibodies brought into question the possibility of other diagnoses such as mixed connective tissue disease and scleroderma, but due to the absence of clinical manifestations such as those observed in systemic sclerosis and Raynaud’s phenomenon, a diagnosis could not be established in accordance with the ACR-EULAR diagnostic criteria [18].
There are currently no standard guidelines for the treatment and management of LAHPS. However, the current mainstay of treatment of patients with LAHPS includes the provision of supportive therapy in the form of red cell transfusions, factor concentrates, and other blood constituents. Furthermore, along with supportive treatments, patients tend to be offered immunosuppressive therapy, including corticosteroids, cyclophosphamide, and other immunosuppressive therapies [19]. In some cases, namely drug-induced and infection-associated LAHPS, the prothrombin deficiency resolved spontaneously with no need for further treatment. However, in a review of 77 patients with LAHPS, 60% of patients received corticosteroid therapy, most commonly with prednisolone at a dose of 1 mg/kg/day. The review reported normalization of PT time and prothrombin levels in most patients. However, in approximately 8% of patients tapering the dose of prednisolone resulted in a transient decrease in prothrombin levels and relapse of LAHPS [15]. Moreover, around 30% of patients received immunosuppressive therapy in the form of cyclophosphamide, azathioprine, or rituximab. The aforementioned immunosuppressive therapies were initiated either as monotherapy or in combination with corticosteroid therapy [15]. Hydroxychloroquine, a disease-modifying agent used in the treatment of SLE, was initiated in 7 cases reported in the literature. Hydroxychloroquine was used in combination with corticosteroids and resulted in a positive response in all patients [15]. Our patient received prednisolone at 1 mg/kg and was initiated on hydroxychloroquine to manage his concurrent SLE diagnosis.
Pseudotumor cerebri is a disease that involves an idiopathic rise in intracranial pressure associated with papilledema in the absence of focal neurological signs or any space-occupying lesions [8]. Pseudotumor cerebri tends to be classically present in young, overweight females [8]. Our patient presented to the Emergency Department 3 months prior to his admission with LAHPS with a background of constant headaches and nausea and was diagnosed with the condition as a result of the lumbar puncture and fundoscopy findings. Pseudotumor cerebri in association with SLE is rare, accounting for about 0.7% of cases [20]. Moussa et al recently published a case series of 8 children who were diagnosed with pseudotumor cerebri and found to have juvenile SLE [21]. The pathogenesis of pseudo-tumor cerebri with SLE is unclear, with the currently proposed mechanisms being an immune-mediated injury to the arachnoid-villi and surrounding structures, thereby reducing cerebrospinal fluid (CSF) reabsorption. The other proposed mechanism is vascular occlusion of the cerebral venous system, resulting in CSF build-up [22,23]. There have been a number of reported cases of pseudotumor cerebri in association with primary antiphospholipid syndrome [24]. Moreover, in a number of these cases, the association was observed in the presence of venous sinus thrombosis [25]. In addition, a meta-analysis of 51 patients observed a significant association between antiphospholipid antibodies and the presence of pseudotumor cerebri [odds ratio (OR) of 4.25 (1.68–12.60)] [26].
Conclusions
We report a case of pseudotumor cerebri with a further presentation of LAHPS in a patient found to have SLE. As both of these associations are exceedingly rare in the presence of SLE, it is essential to recognize them early to initiate adequate management and intervention. As a result of the exceptionally rare incidence of LAHPS and its potential to cause life-threatening bleeding, it is imperative to understand the clinical manifestations of the disease to manage patients. Furthermore, patients with SLE and positive LA antibodies should be closely monitored and observed if presenting with any form of bleeding and hemorrhage. Early recognition of LAHPS often results in initiating early treatment with corticosteroids and immunosuppressant, which provides a good outcome in most patients [15]. Moreover, patients with LAHPS require close follow-up to assess disease progression and treat and manage relapse effectively. Furthermore, pseudotumor cerebri is a rare complication of SLE, which can lead to severe vision defects if left untreated. The presentation of patients with SLE or APLS with symptoms of a raised intracranial pressure such as headaches and nausea should prompt urgent investigation, including fundoscopic examination, CT head, CT venogram, and lumbar puncture, to accurately identify pseudotumor cerebri and provide appropriate interventions to avoid visual defects.
References:
1.. Hoffman MM, Monroe DM, Rethinking the coagulation cascade: Curr Hematol Rep, 2005; 4(5); 391-96
2.. Lancellotti S, Basso M, De Cristofaro R, Congenital prothrombin deficiency: An update: Semin Thromb Hemost, 2013; 39(6); 596-606
3.. Setty YN, Komatireddy GR, Antiphospholipid antibodies in systemic lupus erythematosus and the antiphospholipid syndrome: Front Biosci, 2001; 6; E207-12
4.. Lollar P, Pathogenic antibodies to coagulation factors. Part II. Fibrinogen, prothrombin, thrombin, factor V, factor XI, factor XII, factor XIII, the protein C system and von Willebrand factor: J Thromb Haemost, 2005; 3(7); 1385-91
5.. Triplett DA, Lupus anticoagulants/antiphospholipid-protein antibodies: The great imposters: Lupus, 1996; 5(5); 431-35
6.. Bajaj SP, Rapaport SI, Fierer DS, A mechanism for the hypoprothrombinemia of the acquired hypoprothrombinemia-lupus anticoagulant syndrome: Blood, 1983; 61(4); 684-92
7.. Ieko M, Yoshida M, Naito S, Lupus anticoagulant-hypoprothrombinemia syndrome and similar diseases: Experiences at a single center in Japan: Int J Hematol, 2019; 110(2); 197-204
8.. Friedman DI, Jacobson DM, Diagnostic criteria for idiopathic intracranial hypertension: Neurology, 2002; 59(10); 1492-95
9.. Maloney K, Idiopathic intracranial hypertension as an initial presentation of systemic lupus erythematosus: BMJ Case Rep, 2013; 2013; bcr2013010223
10.. Devreese KMJ, de Groot PG, de Laat B, Guidance from the Scientific and Standardization Committee for lupus anticoagulant/antiphospholipid antibodies of the International Society on Thrombosis and Haemostasis: Update of the guidelines for lupus anticoagulant detection and interpretation: J Thromb Haemost, 2020; 18(11); 2828-39
11.. Aringer M, Costenbader K, Daikh D, 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus: Arthritis Rheumatol, 2019; 71(9); 1400-12
12.. Miyakis S, Lockshin MD, Atsumi T, International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS): J Thromb Haemost, 2006; 4(2); 295-306
13.. Rapaport SI, Ames SB, Duvall BJ, A plasma coagulation defect in systemic lupus erythematosus arising from hypoprothrombinemia combined with antiprothrombinase activity: Blood, 1960; 15; 212-27
14.. Mazodier K, Arnaud L, Mathian A, Lupus anticoagulant-hypoprothrombinemia syndrome: report of 8 cases and review of the literature: Medicine (Baltimore), 2012; 91(5); 251-60
15.. Mulliez SM, De Keyser F, Verbist C, Lupus anticoagulant-hypoprothrombinemia syndrome: Report of two cases and review of the literature: Lupus, 2015; 24(7); 736-45
16.. Zamora-Medina MDC, Hinojosa-Azaola A, Nuñez-Alvarez CA, Anti-RNP/ Sm antibodies in patients with systemic lupus erythematosus and its role in thrombosis: A case-control study: Clin Rheumatol, 2019; 38(3); 885-93
17.. Galli M, Luciani D, Bertolini G, Barbui T, Lupus anticoagulants are stronger risk factors for thrombosis than anticardiolipin antibodies in the antiphospholipid syndrome: A systematic review of the literature: Blood, 2003; 101(5); 1827-32
18.. van den Hoogen F, Khanna D, Fransen J, 2013 classification criteria for systemic sclerosis: An American college of rheumatology/European league against rheumatism collaborative initiative: Ann Rheum Dis, 2013; 72(11); 1747-55
19.. Pilania RK, Suri D, Jindal AK, Lupus anticoagulant hypoprothrombinemia syndrome associated with systemic lupus erythematosus in children: Report of two cases and systematic review of the literature: Rheumatol Int, 2018; 38(10); 1933-40
20.. Roongta R, Chattopadhyay A, Bhattacharyya S, Ghosh A, Pseudotumour cerebri in lupus: Clin Rheumatol, 2021; 40(4); 1661-62
21.. Moussa T, Abdelhak M, Edens C, Pseudotumor cerebri syndrome in children with systemic lupus erythematosus: Case series and review: Pediatr Rheumatol Online J, 2022; 20(1); 29
22.. Kim JM, Kwok SK, Ju JH, Idiopathic intracranial hypertension as a significant cause of intractable headache in patients with systemic lupus erythematosus: A 15-year experience: Lupus, 2012; 21(5); 542-47
23.. Green L, Vinker S, Amital H, Pseudotumor cerebri in systemic lupus erythematosus: Semin Arthritis Rheum, 1995; 25(2); 103-8
24.. Leker RR, Steiner I, Anticardiolipin antibodies are frequently present in patients with idiopathic intracranial hypertension: Arch Neurol, 1998; 55(6); 817-20
25.. Mokri B, Jack CR, Petty GW, Pseudotumor syndrome associated with cerebral venous sinus occlusion and antiphospholipid antibodies: Stroke, 1993; 24(3); 469-72
26.. Kesler A, Kliper E, Assayag EB, Thrombophilic factors in idiopathic intracranial hypertension: A report of 51 patients and a meta-analysis: Blood Coagul Fibrinolysis, 2010; 21(4); 328-33
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133