17 June 2023: Articles 
A 78-Year-Old Man with Chronic Kidney Disease and Monoclonal Gammopathy Who Developed Post-Transplant C3 Glomerulopathy – Recurrence or De Novo? A Case Report and Literature Review
Challenging differential diagnosis, Rare coexistence of disease or pathology
María Carmen Ruiz-FuentesDOI: 10.12659/AJCR.939726
Am J Case Rep 2023; 24:e939726






Abstract
BACKGROUND: The incidence of glomerular disease recurrence in kidney transplant patients varies according to type of glomerulopathy; therefore, it is important to know the primary chronic kidney disease etiology. C3 glomerulopathy (C3G) is characterized by deposits of C3 in immunofluorescence and its pathogeny is based on the dysregulation of the alternative complement pathway. C3G has a high recurrence rate and, given its low prevalence, only case series have been published. A higher rate of recurrence and a more aggressive course have been described in association with monoclonal gammopathy (MG).
CASE REPORT: We describe the case of a 78-year-old man with chronic kidney disease of unknown etiology (no significant proteinuria) and monoclonal IgGl gammopathy with low risk of progression, who received a kidney transplant, presenting accelerated deterioration of kidney function. Histopathology showed predominant C3 deposits in immunofluorescence, compatible with C3 glomerulonephritis (C3GN). He was treated with eculizumab during 4 weeks while the study was completed. The response to treatment was not favorable and the patient remained in the dialysis program.
CONCLUSIONS: Further studies are needed to explain the pathogenic mechanisms of complement alternative pathway dysregulation mediated by monoclonal component in patients with C3GN and MG. Patients older than 50 years who are on a waiting list for kidney transplantation should have an MG detection study. The information provided to patients with MG on a waiting list for kidney transplantation should include not only the possibility of hematologic progression but also the recurrence/de novo appearance of associated kidney pathology.
Keywords: Kidney Transplantation, Paraproteinemias, Recurrence, Male, Humans, Aged, Complement C3, Renal Dialysis, Glomerulonephritis, monoclonal gammopathy of undetermined significance, Renal Insufficiency, Chronic, Glomerulonephritis, Membranoproliferative
Background
Recurrence of primary disease in kidney transplantation occurs in 20% of patients, and this causes graft loss in 8.4% of cases 10 years after transplantation [1]. Despite this recurrence, transplantation remains the best option for patients with stage 5 chronic kidney disease (CKD) when there are no contraindications. The 2020 Spanish Society of Nephrology Registry of CKD report shows that 18% of incident patients in substitutive renal therapy had unknown etiology of their renal disease. According to a recent study, the percentage of patients with unknown etiology or glomerulonephritis without histologic diagnosis in CKD, in which primary disease recurs after kidney transplantation, is low [2].
The incidence of recurrence in patients with known primary glomerular pathology varies depending on the pathology. In the specific case of C3 glomerulopathy (C3G), recurrence is high. The clinical features of this entity are proteinuria, usually nephrotic, microhematuria, and a variable degree of renal failure. C3G is characterized by deposits of C3 in immunofluorescence and its pathogeny is based on the dysregulation of the alternative complement pathway. There are 2 entities of C3G, C3 glomerulonephritis (C3GN) and dense deposits disease (DDD), in which presence of highly electron-dense deposits within the glomerular basement membrane are observed by electronic microscopy. There is a low incidence (1–3 cases per million a year) of this particular disease and this is based mainly on case series; but studies of larger groups of patients describe recurrences of 67–84% in C3GN, and up to 100% in dense deposits disease (DDD) [1]. Monoclonal gammopathy (MG) associated with C3G expands the concept, naming this entity “monoclonal gammopathy of renal significance” (MGRS), presenting worse outcomes [3,4]. De novo C3G is even less described in the literature. The management of C3G is not well defined; among the proposed treatments is eculizumab, a membrane attack complex blocker [5].
This report is of a 78-year-old man with chronic kidney disease and monoclonal gammopathy who developed post-transplant C3 glomerulonephritis with lack of response to eculizumab.
Case Report
Our patient was a 78-year-old man with a history of CKD of unknown etiology and proteinuria of 0.5–0.8 gr in 24 h. His complement test result pretransplant was unknown (he came from another unit of Nephrology). His glomerular filtration rate decreases over about 3 years, and then he began a hemodialysis program. Other characteristics of his clinical history were: high blood pressure of 10 years of evolution, IgGl uncertain significance MG of low progression risk, without fat infiltration or bone marrow (PET-CT without hypermetabolic focus that could suggest macroscopic myeloma affectation), chronic thrombocytopenia, prostatic hypertrophy, hematuria without urologic significance, lipid abnormalities, and non-smoking.
He received a kidney transplant from a deceased donor (a 70-year-old woman and a cold ischemia time of 12 h), sharing blood group and 1 HLA compatibility (DR7). Basiliximab, mycophenolate mofetil, and methylprednisolone were the induction treatment. Tacrolimus was introduced on day +5.
Since transplantation, the patient did not present clinical symptoms, and had no radiology or blood test that indicated bacterial or viral active infection. There was no isolation of other microorganism other than
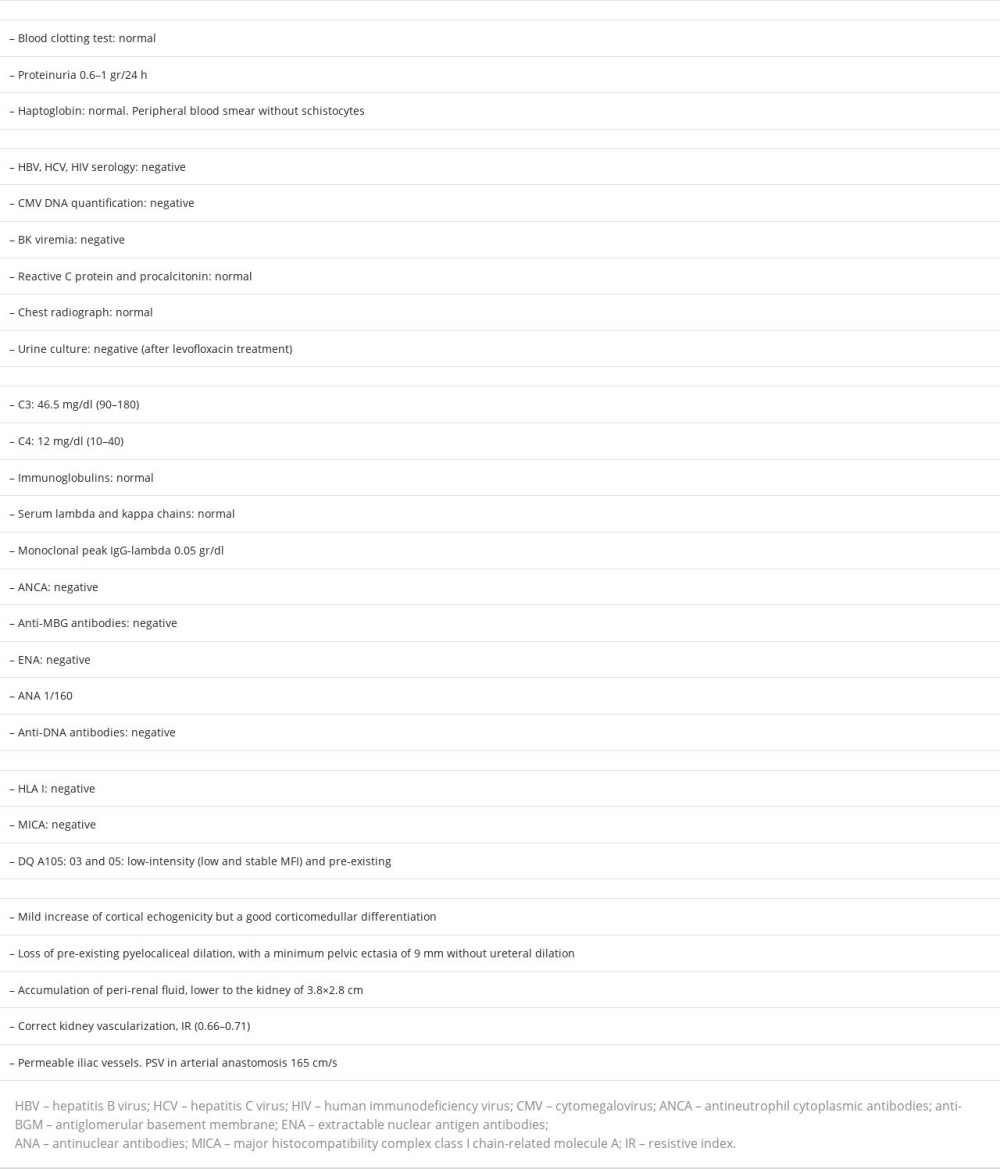
Eculizumab was administered on day +41 post-transplant (creatinine 8.68 mg/dl on day +39) due to compatible dates with high probability of C3G and the early and rapid worsening of renal function, with substitutive renal therapy required (the patient previously received meningococcus vaccination and treatment with amoxycillin-clavulanic). A hematological study was requested in relation to MG, leucopenia, and thrombocytopenia to evaluate the possibility of treatment for that pathology that could affect and slow the unfavorable evolution of renal function, but this option was not considered suitable at that time. After 4 doses of eculizumab (900 mg/weekly, 4 weeks), the patient did not show any indication of clinical or blood test improvement (creatinine 10.1 mg/dl, 9.22 mg/dl, 8.28 mg/dl on days +43, +49, +64 respectively), so he continued in the hemodialysis program.
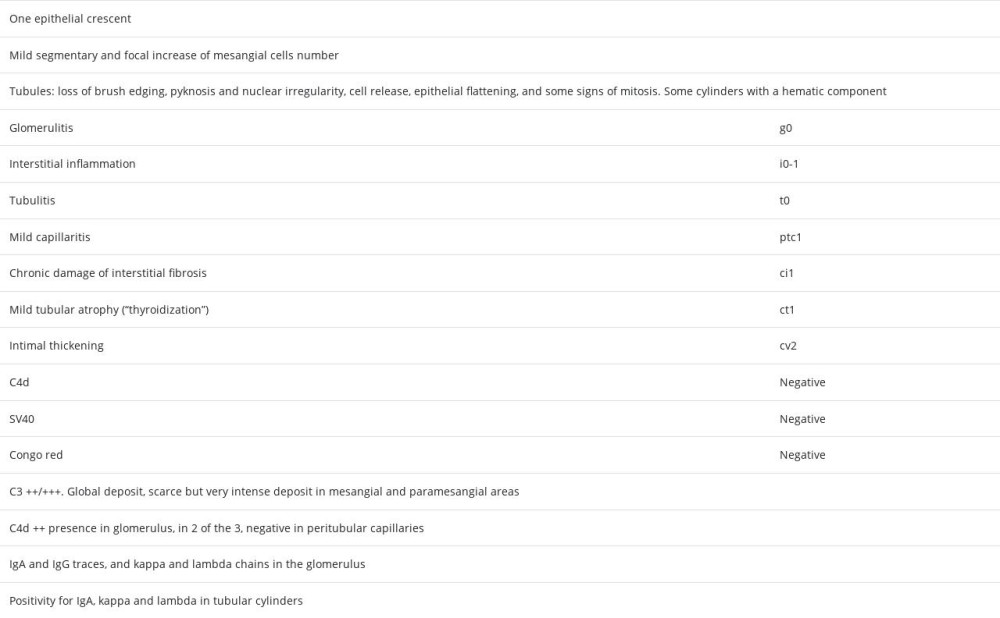
Other studies carried out included: complement genetic test without alterations in the genes CFH, CD46, THBD, CFI, C3, CFHR1, and CFHR5; negative C3 nephritic factor (C3NeF), normal anti-FH antibodies; electronic microscopy in renal biopsy that described podocytes with large areas of preserved pedicels and some focal pedicellar fusion, microvillous transformation, electron-dense deposits isolated in the mesangial and paramesangial areas, and there were no signs of glomerulitis or transplant glomerulopathy (Figure 3). The absence of dense deposits in glomerular basement membrane focused our diagnosis on G3GN.
Discussion
This case shows the importance of knowing the CKD etiology and patient comorbidity before kidney transplantation, not only for treatment options but for educating the patient in the pretransplant period. The recurrence of the primary disease in renal transplantation is an important cause of graft loss. Native kidney biopsy is not performed in different cases where there is suspected glomerular pathology due to several reasons (eg, comorbidity, patient refusal, advanced CKD), and the etiology of CKD is defined as presumed chronic glomerulonephritis without being biopsy-proven. In other cases, it is impossible to determine the etiology of CKD in the absence of significant proteinuria or identifiable risk factors. Both groups could show variable incidence according to several registries, which may vary between 10% to 25% in some cases. A study based on the Australia and New Zealand Dialysis and Transplant registry [2] compared the recurrence of kidney disease in patients with chronic glomerulonephritis (CGN) proven by biopsy prior to transplantation with the recurrence of those with CGN not proven by biopsy and of unknown etiology. The second group presented lower risk of post-transplant glomerular disease than the first one. In any case, it is important to know the histological diagnosis in patients with suspected CGN to be able to inform them of the primary disease recurrence risk after kidney transplantation.
The definition of C3G is relatively new, with its 2 histologic subtypes, C3GN and DDD [6,7]. This pathology is considered a rare disease, so its description is limited to case series and retrospective registries. The pathogeny of the disease is related to the dysregulation of the alternative complement pathway in its fluid phase and in the glomerular environment. Hyperactivation of the complement produces the deposit of the C3 fraction and its excision products in the glomerulus. Although the clinical presentation is variable, the most frequent pattern is significant proteinuria with relatively maintained renal function, but it can present proteinuria <1 g/24 h both in the primary disease and in recurrence after transplantation [2,3]. The histological pattern is typified by positive isolated or predominant deposits of C3 in IF. Electronic microscopy in C3GN shows electron-dense deposits located in the mesangial area and subendothelial side of the glomerular basement membrane [8].
Several series showed a strong association of C3G with MG in patients over 50 years of age [7,9]. The monoclonal component action would not be caused by deposits in the kidney. According to classic reference studies, the monoclonal component would act as an inhibitor of complement-regulating proteins; for example, the dimeric monoclonal l light chain would be a nephritogenic factor that would act as an antibody against several factors such as factor H [9,10], and would activate the alternative complement pathway.
Chauvet et al [11] made a comparison between complement dysregulation mechanisms in patients with C3G plus monoclonal immunoglobulin (Ig) and patients without monoclonal peak, and found that in the monoclonal Ig group, only 7% had complement genetic alterations, while 27% of the other group without monoclonal Ig had them. C3NeF was found in 7% in the first group vs 45% in the second. The C5 nephritis factor was negative in the first group, anti-factor H antibodies were low and similar in both groups, and anti-CR1 was found in 27% of the patients with monoclonal Ig but undetectable in the other group. This study showed agreement between both isotypes of the light and heavy chain of the monoclonal Ig and the complement anti-proteins antibodies in only 3 of 13 patients, which suggests that the reactivity of the complement anti-proteins is not mediated by the Ig monoclonal component. This argument outlines a new hypothesis, namely that monoclonal Ig acts as a complement-activating surface. This scenario has not yet been explored and more studies are needed to explain the pathogenic mechanisms related to dysregulation of the alternative complement pathway mediated by a monoclonal component.
The recurrence of C3GN is high in renal transplantation, and is described in the main series of the Mayo Clinic [3] and Columbia University [12], with an incidence of 67–84%, showing that recurrence is earlier and more aggressive in patients with MG and C3G.
In 2 series (Mayo Clinic and a French series) [13,14] in which the repercussion of the hematologic process treatment on renal prognosis was studied, the recurrence of C3G in patients with MG was 100% after transplantation.
In the case of the appearance of de novo C3G, studies are even scarcer [15,16]. A Spanish case series described the association of C3G de novo and MG [17]. In 2 of the 3 cases, the presentation of C3G was in patients with unknown primary disease, without proteinuria, and MG was diagnosed after the histological diagnosis of C3G, but there was no information on whether MG existed before or not.
We report the case of a 78-year-old man with CKD of unknown etiology (proteinuria 0.5–08 g/24 h), with a relatively fast progression to hemodialysis (3 years), and an IgGl MG of low probability of progression, who presented an early and fast progressive worsening of renal function after transplantation, low C3 fraction, and histology compatible with C3GN (dismissing a post-infectious etiology), without response to eculizumab and requiring hemodialysis. Are we facing a de novo or recurrent C3GN? The evolution of the primary disease until hemodialysis and the fast deterioration of renal function in early after transplant point toward a C3GN recurrence. We could relate the renal pathology to MG because the genetic analysis of complement was negative. The available functional test of complement in our case were C3NeF and anti-FH antibodies, and they were negative. In the published series, there is no pattern of the presence of complement antiprotein antibodies, and even levels of the C3 and C4 fractions are not characteristic [1,18]. Unfortunately, other antibodies (eg, anti-FI, antiCR1) could not be determined in this case, nor was C3 convertase activity or C5b9.
In the case of de novo C3G associated with MG, no publications were found that clearly described the absence of MG before transplantation.
In the present case, treatment with eculizumab was begun due to the rapid deterioration of renal function, with the aim of trying to stabilize the progression and evaluate the possibility of hematological treatment [19]. That treatment was dismissed due to poor outcome, lack of indication according to hematology criteria (stable monoclonal peak), and significant leucopenia and thrombocytopenia.
Two recent series described the relationship of kidney disease with MG and the evolution of both, depending on treatment. In a French series [14] of 201 patients with C3G, 60 presented MG. The mean renal survival rate was higher in patients who had received chemotherapy than in those who had conservative treatment. In the chemotherapy group, the renal survival was higher in those who achieved complete hematological response, which underlines the relevance of a focused therapy on the MG specific clone. Four patients were recipients of a kidney transplant (without complete hematological remission), and all of them had a C3G recurrence (3–12 months).
The second series, from the Mayo Clinic, studied several cases of renal-significant MG in patients who received kidney transplantation [13]. In the C3G group, there were 5 patients, 3 of them were diagnosed with C3G after post-transplant recurrence (the native kidney biopsies were reviewed), none of them presented genetic complement alterations, all recurred after kidney transplant (mean of 70 days), and 3 of them progressed to myeloma after kidney transplantation. Regarding treatment, 3 of the patients were not treated before transplantation. Only the patient who achieved complete remission of hematologic pathology had favorable renal and total survival after recurrence of C3G. There are few reported cases, and more studies are necessary to evaluate the effect of hematological treatment on the evolution of C3G in kidney transplantation.
Conclusions
Our experience with this case and the literature review on C3G associated with MG show the need for further studies that could explain the pathogenic mechanisms of complement alternative pathway dysregulation mediated by monoclonal component, and the hematologic treatment effect on kidney evolution.
In our opinion, patients older than 50, on a waiting list for kidney transplantation, and with unknown CKD etiology should have a complement determination and MG detection study.
We believe that the information on patients with MG on a waiting list for kidney transplantation should include not only the possibility of hematologic progression but also the recurrence/de novo appearance of associated kidney pathology.
References:
1.. Chadban SJ, Ahn C, Axelrod DA, KDIGO clinical practice guideline on the evaluation and management of candidates for kidney transplantation: Transplantation, 2020; 104(4 Suppl. 1); S11-S103
2.. Lim WH, Wong G, McDonald SP, Long-term outcomes of kidney transplant recipients with end-stage kidney disease attributed to presumed/ advanced glomerulonephritis or unknown cause.: Sci Rep, 2018; 8(1); 9021
3.. Zand L, Lorenz EC, Cosio FG, Fervenza FC, Clinical findings, pathology, and outcomes of C3GN after kidney transplantation: Am Soc Nephrol, 2014; 25(5); 1110-17
4.. Amaador K, Peeters H, Minnema MC, Monoclonal gammopathy of renal significance (MGRS) histopathologic classification, diagnostic workup, and therapeutic options: Neth J Med, 2019; 77(7); 243-54
5.. González Suárez ML, Thongprayoon C, Hansrivijit P, Treatment of C3 glomerulopathy in adult kidney transplant recipients: A systematic review: Med Sci (Basel), 2020; 8(4); 44
6.. Pickering MC, D’Agati VD, Nester CM, C3 glomerulopathy: Consensus report: Kidney International, 2013; 84; 1079-89
7.. Lloyd IE, Gallan A, Huston HK, C3 glomerulopathy in adults: A distinct patient subset showing frequent association with monoclonal gammopathy and poor renal outcome: Clin Kidney J, 2016; 9(6); 794-99
8.. Caravaca-Fontán F, Lucientes L, Cavero T, Praga M, Update on C3 glomerulopathy: A complement-mediated disease: Nephron, 2020; 144(6); 272-80
9.. Meri S, Koistinen V, Miettinen A, Activation of the alternative pathway of complement by monoclonal lambda light chains in membranoproliferative glomerulonephritis: J Exp Med, 1992; 175; 939-50
10.. Jokiranta TS, Solomon A, Pangburn MK, Nephritogenic lambda light chain dimer: A unique human miniautoantibody against complement factor H: J Immunol, 1999; 163; 4590-96
11.. Chauvet S, Roumenina LT, Aucouturier P, Both monoclonal and polyclonal immunoglobulin contingents mediate complement activation in monoclonal gammopathy associated-C3 glomerulopathy: Front Immunol, 2018; 2(9); 2260
12.. Regunathan-Shenk R, Avasare RS, Ahn W, Kidney transplantation in C3 glomerulopathy: A case series.: Am J Kidney Dis, 2019; 73(3); 316-23
13.. Heybeli C, Alexander MP, Bentall AJ, Kidney transplantation in patients with monoclonal gammopathy of renal significance (MGRS) – associated lesions: A case series: Am J Kidney Dis, 2022; 79(2); 202-16
14.. Chauvet S, Frémeaux-Bacchi V, Petitprez F, Treatment of B-cell disorder improves renal outcome of patients with monoclonal gammopathy-associated C3 glomerulopathy: Blood, 2017; 129(11); 1437-47
15.. Lim WH, Shingde M, Wong G, Recurrent and de novo glomerulonephritis after kidney transplantation: Front Immunol, 2019; 10; 1944
16.. Nahm JH, Song SH, Kim YS, De novo C3 glomerulonephritis in a renal allograft: Ultrastruct Pathol, 2016; 40; 112-15
17.. Serra N, Facundo C, Canal C, Three cases of monoclonal gammopathy of renal significance after kidney transplantation. De novo C3 glomerulopathy: Nefrología, 2019; 39(2); 198-201
18.. Bridoux F, Desport E, Frémeaux-Bacchi V, Glomerulonephritis with isolated C3 deposits and monoclonal gammopathy: A Fortuitous Association?: Clin J Am Soc Nephrol, 2011; 6(9); 2165-74
19.. Moog P, Jost PJ, Büttner-Herold M, Eculizumab as salvage therapy for recurrent monoclonal gammopathy-induced C3 glomerulopathy in a kidney allograft: C Nephrol, 2018; 19(1); 106
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133