28 January 2024: Articles 
A Case of Catastrophic Antiphospholipid Syndrome with Acute Multiorgan Thrombosis and Concerns for Re-Emergence
Rare disease
Sushmita MittalDOI: 10.12659/AJCR.942339
Am J Case Rep 2024; 25:e942339






Abstract
BACKGROUND: Catastrophic antiphospholipid syndrome (CAPS) is a rare, life-threatening form of antiphospholipid syndrome characterized by widespread thrombotic complications leading to multiorgan ischemia and failure. Although there are no standard treatment guidelines for CAPS, it often involves triple therapy with anticoagulation, corticosteroids, and plasma exchange. Recently, biologics such as rituximab and eculizumab have also shown promise as potential new therapies for CAPS, as observed in our case.
CASE REPORT: We describe a 59-year-old female patient who presented with altered mental status and diffuse weakness. Imaging studies revealed multiorgan thrombosis along with thrombocytopenia that markedly improved with plasma exchange therapy, steroids, and a heparin drip. While the exact etiology of CAPS remained unknown, it was likely precipitated by her warfarin discontinuation and confirmed Haemophilus influenzae infection. The patient’s hospital course was complicated by hemorrhagic shock after a renal biopsy, followed by an acute drop in thrombocytopenia and new embolic infarcts in the brain that raised concern for CAPS re-emergence. To address the refractory nature of her condition, the patient underwent a trial of rituximab, which remarkably improved her clinical picture and platelet count by an 8-fold increase within 1 week.
CONCLUSIONS: This case highlights the importance of early recognition and diagnosis of catastrophic antiphospholipid syndrome, a true rheumatological emergency that requires aggressive treatment to prevent irreversible complications. Our patient’s presentation and response to treatment also underscores the complexity of managing CAPS and the use of newer biological therapies in refractory cases.
Keywords: Antiphospholipid Syndrome, rituximab, Thrombosis, Female, Humans, Middle Aged, Heparin, Thrombocytopenia
Background
Antiphospholipid syndrome (APS), also known as Hughes syndrome, is an autoimmune condition that involves arterial and venous thromboembolism and/or recurrent pregnancy loss due to the presence of antiphospholipid antibodies. APS can occur as a primary condition or can be secondary to other autoimmune conditions, such as systemic lupus erythematosus (SLE) [1].
Catastrophic APS (CAPS), also known as Asherson’s syndrome, is a rare, life-threatening form of APS with a mortality rate of about 50% [2]. CAPS is defined by acute widespread thrombosis in several vascular beds leading to multiorgan ischemia and failure. There are approximately 805 cases of CAPS that have been reported around the world, about 1% occurring in patients with known APS. Due to the rarity of this syndrome, an international registry of patients with CAPS was created by the European Forum on Antiphospholipid Antibodies in 2000 to help establish diagnostic criteria and management of CAPS [3]. We present our case to highlight the acute multiorgan thrombosis, severe thrombocytopenia, and hemorrhagic complications that can be seen in the initial presentation of CAPS.L1
Case Report
We present a case of a 59-year-old woman with a past medical history of systemic lupus erythematosus, antiphospholipid syndrome with a history of recurrent deep vein thromboses (DVTs) managed with warfarin, multiple miscarriages, hyper-tension, chronic kidney disease stage 3, seizures, and a subdural hematoma. She was transferred to our institution for worsening encephalopathy, requiring a higher level of care in the intensive care unit with intubation for airway protection. The patient’s husband reported she had 1 day of altered mental status and profound migratory extremity weakness. He also mentioned the patient had stopped taking warfarin 2 weeks before this incident due to a “supratherapeutic international normalized ratio (INR).”
On presentation, she was afebrile but tachycardic (110 beats per minute) and hypertensive (200/100 mmHg). On examination, she was somnolent, not following commands, had diffuse petechiae and ecchymosis, and significant non-pitting edema on bilateral upper extremities (BUE) with 1+ pitting edema on the right lower extremity (RLE). Initial laboratory workup revealed pancytopenia along with elevated creatinine and normal blood urea nitrogen (Table 1). She also tested positive for
An upper-extremity venous ultrasound revealed acute occlusive DVT in the right brachial vein with distal non-occlusive extension into the right subclavian vein (Figures 1, 2) along with acute occlusive thrombosis of the left basilic vein, which was likely superficial thrombophlebitis. A lower-extremity venous ultrasound revealed chronic-appearing non-occlusive DVT involving the right common femoral vein and extending distally to the calf. A chest CT revealed a non-occlusive thrombus in the superior vena cava (Figure 3). An abdomen magnetic resonance imaging (MRI) revealed acute DVTs in the right common iliac, external iliac, and internal iliac veins (Figure 4). A head CT angiogram revealed many areas of luminal narrowing followed by a normal caliber lumen-like “beads on a string” appearance seen in bilateral branches of the posterior cerebral artery (PCA), middle cerebral artery (MCA), and anterior cerebral artery (ACA) with initial concerns of lupus vasculitis; however, these findings were not seen on a brain magnetic resonance angiography (MRA) that was performed 2 days after. A brain MRI also revealed several lacunar infarcts within the left cerebellar hemisphere, left pons, and left basal ganglia (Figures 5, 6). An echocardiogram was also performed, which revealed no abnormal findings.
Further workup for her diffuse thrombosis in the setting of thrombocytopenia was performed and revealed an elevated D-dimer (5976 ng/mL), fibrinogen (770 mg/dL), partial thromboplastin time (66 seconds), and prothrombin time (14.8 seconds) with a normal INR (1.2). She also had a negative double-stranded deoxyribonucleic acid antibody (dsDNA), positive antinuclear antibody (ANA) with a titer of 1: 1280, and the presence of immunoglobulin (Ig) A, IgG, and IgM beta-2 glycoprotein-1 antibodies, IgA/IgG/IgM anticardiolipin antibodies, and lupus anticoagulant. Complement levels were normal, including C3 level (98 mg/dL) and C4 level (17 mg/dL), anti-platelet factor 4 (PF4) assay testing was negative, and a peripheral blood smear did not reveal any schistocytes (Table 2).
Her diffuse thrombosis, thrombocytopenia, and positive antiphospholipid antibodies raised immediate concern for catastrophic antiphospholipid syndrome. Although the exact etiology of CAPS was undetermined, it was thought to have been precipitated by her discontinuation of warfarin along with her underlying
Within days of triple therapy consisting of anticoagulation, corticosteroids, and plasma exchange, the patient’s neurological function and platelet count significantly improved (stabilized to 100×109/L), leading to her transfer out of the intensive care unit. This improvement was short-lived, however, after she underwent a renal biopsy and subsequently developed a large left subcapsular renal hemorrhage, leading to hemorrhagic shock. Further workup after this event revealed an acute drop in platelet count (19×109/L) despite high-dose oral steroids, which raised concern for the re-emergence of CAPS. Repeat brain MRI revealed scattered small embolic cortical and sub-cortical infarcts in bilateral frontal and parietal lobes, and renal biopsy revealed antiphospholipid nephropathy with acute-on-chronic thrombotic microangiopathy changes. All these findings supported the suspicion of CAPS re-emergence.
Anticoagulation was held in the setting of her recent hemorrhage, and despite receiving a high dose of oral prednisone (100 mg), her platelet count did not increase. Due to refractory CAPS, the patient was trialed on 2 doses of rituximab 1 g IV infusions, given 1 week apart, which significantly improved her clinical status and platelet count (from 19×109/L to 155×109/L) within 1 week. During this time, she also underwent an additional 5 days of PLEX and her platelet count remained stabilized on discharge (144×109/L). Anticoagulation was also restarted, eventually transitioning to oral warfarin on discharge.
Discussion
Antiphospholipid syndrome (APS) is an autoimmune disorder characterized by hypercoagulability that is associated with a high risk of arterial and venous thrombosis and recurrent miscarriages. It can present either as an isolated disorder (primary APS) or occur secondary to other autoimmune conditions like systemic lupus erythematosus [1]. Catastrophic antiphospholipid syndrome (CAPS) is a rare, life-threatening variant of APS associated with widespread vascular thrombosis, often leading to rapid-onset multiorgan failure. Approximately 1% of APS patients develop CAPS, which has a mortality rate of up to 30%. Like many autoimmune conditions, women account for 70% of CAPS cases [2].
CAPS is frequently triggered by infections, surgical procedures, malignancies, trauma, systemic lupus erythematosus flares, pregnancy complications, and discontinuation of anticoagulation [4]. Although the exact cause of CAPS in our patient was unknown, it is likely that her CAPS was precipitated by a combination of discontinuing her warfarin and a confirmed
A definitive diagnosis of CAPS requires the following 4 criteria to be met: 1) thrombosis of 3 or more organs or tissues; 2) manifestations occurring in less than 1 week; 3) histopathological evidence of small vessel occlusion; and 4) confirmed presence of antiphospholipid antibodies on 2 occasions 6 weeks apart [5]. Our patient met the first 3 criteria for diagnosis, which indicated a probable diagnosis of CAPS. She presented with acute deep vein thromboses in the right brachial vein, right subclavian vein, right common iliac vein, right external iliac vein, and right internal iliac vein, along with non-occlusive thrombosis in the superior vena cava and right common femoral vein. She also had thrombotic occlusions of the left kidney and several cerebrovascular accidents within 1 week. Our patient also tested positive for all 3 antiphospholipid antibodies, and renal biopsy revealed histopathologic evidence of CAPS with findings of antiphospholipid nephropathy with acute-on-chronic thrombotic microangiopathy. Unfortunately, our patient did not return to the rheumatology clinic after discharge, so repeat antiphospholipid antibodies could not be drawn 6 weeks after the initial study to confirm a definitive diagnosis of CAPS.
Diagnosing CAPS accurately also involves ruling out other differential diagnoses that can also present with multiple thromboses and concurrent thrombocytopenia. Some of the major ones include disseminated intravascular coagulation (DIC), heparin-induced thrombocytopenia (HIT), thrombotic thrombocytopenic purpura (TTP), and malignant hemopathies [6]. HIT was excluded in our patient’s case since she presented with diffuse thrombosis and simultaneous thrombocytopenia prior to anticoagulation use and tested negative for the presence of heparin-PF4 antibodies [7]. There was a low suspicion of TTP as our patient’s peripheral blood smear did not reveal elevated levels of schistocytes. DIC was also unlikely in our patient, who presented with large-vessel thrombosis and high fibrinogen levels, as it typically presents with low fibrinogen levels and thrombosis affecting small vessels [8].
Typical laboratory findings in CAPS include thrombocytopenia, antiphospholipid antibodies (beta-2 glycoprotein-1 antibody, anticardiolipin antibody, lupus anticoagulant), and antinuclear antibodies, all of which were seen in our patient. Thrombocytopenia in CAPS results from the rapid clot formation, which depletes platelet levels. Additionally, some patients with CAPS can have low levels of C3 and C4 due to activation of the complement system. One study performed using the CAPS Registry revealed that out of the 73 patients studied, 42 patients (58%) were found to have low levels of C3 and C4. Furthermore, low levels were more commonly seen in patients with SLE. Although our patient had also had a history of SLE, her C3 and C4 were normal, which we believe is because she was not having an active flare of SLE. It is theorized that complement activation is one of the mechanisms by which antiphospholipid antibodies in CAPS can induce coagulation and cellular injury, although the exact relationship remains unknown [9].
Clinical presentation of CAPS varies based on the different organ system involved. Our patient presented with involvement of the kidneys, central nervous system (CNS), and skin. Renal involvement is the most common and presents as hypertension, proteinuria, hematuria, and acute renal failure, which were seen in our patient. This occurs due to the occlusion of multiple small vessels in the kidney by platelet-rich thrombi presenting as thrombotic microangiopathy, a common histo-pathological lesion in CAPS that was also seen in our patient’s kidney biopsy. CNS involvement can present as hypertensive or ischemic encephalopathy, stroke, and cerebral venous thrombosis that can cause altered mental status. Our patient initially presented with altered mental status, which could be multi-factorial due to the several lacunar infarcts that were seen on brain MRI on admission along with metabolic encephalopathy. Our patient also presented with cutaneous manifestations of CAPS, including petechiae and ecchymosis [10].
Currently, there are no standard treatment guidelines for patients with CAPS. It often involves triple therapy with anticoagulation, corticosteroids, and plasma exchange. Data from the first 250 patients in the CAPS Registry revealed that this combination of treatment increased survival rates up to 78%. Unfractionated heparin is the main anticoagulation treatment used in CAPS, which is maintained for 7 to 10 days and then switched to oral anticoagulation therapy like warfarin. Glucocorticoids are used as anti-inflammatory agents to treat the systemic inflammatory response that occurs in CAPS. Plasma exchange is used to remove antiphospholipid antibodies and cytokines from plasma [11].
Recent studies have also demonstrated the effectiveness of IV infusions of rituximab and eculizumab, both monoclonal antibodies, as treatments for refractory cases of CAPS. Rituximab has shown benefits against CAPS by blocking the CD20, a B cell antigen, which decreases the formation of antibodies, including antiphospholipid antibodies that increase risk of thrombotic complications. According to a study performed using the CAPS Registry in May 2013, 15 out of 20 people treated with rituximab had successfully recovered from their CAPS episode without having new episodes of thrombosis, indicating a recovery rate of 75% following rituximab administration [12]. Eculizumab works by inhibiting the terminal pathway of the complement system (C5b-9 membrane attack complex), which is known to contribute to the thrombotic manifestations of CAPS. Use of Eculizumab is also supported by another systematic review done using the CAPS Registry, where 29 out of 39 patients (74.4%) treated with eculizumab recovered from CAPS [13].
We use our case to highlight another instance where rituximab can be beneficial in a refractory case of CAPS. Our patient was initially started on triple therapy, which significantly improved her neurological function and platelet count (from 36×109/L to 100×109/L). Unfortunately, her hospital course was complicated by hemorrhagic shock after undergoing a renal biopsy. Further laboratory testing revealed an acute drop in platelet count down to 19×109/L, and repeat brain MRI revealed new scattered small embolic cortical and subcortical infarcts in bilateral frontal and parietal lobes, all concerning for CAPS re-emergence. Despite increasing her oral prednisone dose (100 mg), her platelet count did not improve. Due to refractory CAPS, our patient was ultimately trialed on 2 doses of rituximab 1 g IV infusion given 1 week apart. This markedly improved her clinical picture and platelet count by an 8-fold increase within 1 week and allowed for sustained remission.
Conclusions
This case emphasizes the importance of recognizing and diagnosing CAPS early, as it remains a life-threatening condition with a high mortality rate. The low incidence of CAPS poses significant challenges in conducting randomized studies to establish definitive treatment guidelines. While recent studies using drugs such as rituximab and eculizumab have shown promise in treating refractory cases of CAPS, the exact criterion for administering these therapies continues to remain uncertain. Further research efforts are essential to enhance our understanding of CAPS and explore more effective therapeutic options. By sharing this case, we aim to contribute valuable insight into managing complex and refractory cases using both conventional and newer biological therapies, such as rituximab. Achieving a diagnosis of CAPS can be challenging due to the various clinical presentations of the syndrome, along with its similarities with other conditions like DIC, TTP, and HIT. This case report adds to the limited knowledge available about CAPS to improve patient outcomes and raise awareness about this rheumatological emergency within the medical community.
Figures
References:
1.. Fischer MJ, Rauch J, Levine JS, The antiphospholipid syndrome: Semin Nephrol, 2007; 27; 35-46
2.. Velásquez-Rimachi V, Palma-García L, Pacheco-Barrios K, Successful treatment of catastrophic antiphospholipid syndrome with therapeutic plasma exchange: Hematol Transfus Cell Ther, 2020; 42; 287-91
3.. Ponce A, Rodríguez-Pintó I, Espinosa G, CAPS Registry Project Group/ European Forum on Antiphospholipid Antibodies (supplementary material 3). Pulmonary involvement in catastrophic antiphospholipid syndrome: A descriptive analysis from the “CAPS Registry”: Semin Arthritis Rheum, 2023; 63; 152265
4.. Roberts JR, Finger DR, Catastrophic antiphospholipid syndrome; A “CATASTROPHIC” case of systemic lupus erythematosus: Hawaii Med J, 2004; 63; 362-64
5.. Lee HA, Kim SE, Jung DW, Small bowel necrosis associated with catastrophic antiphospholipid syndrome: A case report: Korean J Gastroenterol, 2021; 77; 294-99
6.. Kitchens CS, Erkan D, Brandão LR, Thrombotic storm revisited: Preliminary diagnostic criteria suggested by the thrombotic storm study group: Am J Med, 2011; 124; 290-96
7.. Warkentin TE, Maurer BT, Aster RH, Heparin-induced thrombocytopenia associated with fondaparinux: N Engl J Med, 2007; 356; 2653-55
8.. Asherson RA, Espinosa G, Cervera R, Disseminated intravascular coagulation in catastrophic antiphospholipid syndrome: Clinical and haematological characteristics of 23 patients: Ann Rheum Dis, 2005; 64; 943-46
9.. Ponce A, Rodríguez-Pintó I, Basauli JM, The clinical significance of low complement levels in patients with catastrophic antiphospholipid syndrome: A descriptive analysis of 73 patients from the “Catastrophic antiphospholipid syndrome registry”: Lupus, 2022; 31; 1218-25
10.. Nayer A, Ortega LM, Catastrophic antiphospholipid syndrome: A clinical review: J Nephropathol, 2014; 3; 9-17
11.. Rodriguez-Pinto I, Espinosa G, Cervera R, Catastrophic antiphospholipid syndrome: The current management approach: Best Pract Res Clin Rheumatol, 2016; 30; 239-49
12.. Berman H, Rodríguez-Pintó I, Cervera R, Rituximab use in the catastrophic antiphospholipid syndrome: Descriptive analysis of the CAPS registry patients receiving rituximab: Autoimmun Rev, 2013; 12; 1085-90
13.. López-Benjume B, Rodríguez-Pintó I, Amigo M, Eculizumab use in catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis from the “CAPS Registry.”: Autoimmunity Reviews, 2022; 21; 103055
Figures
Tables
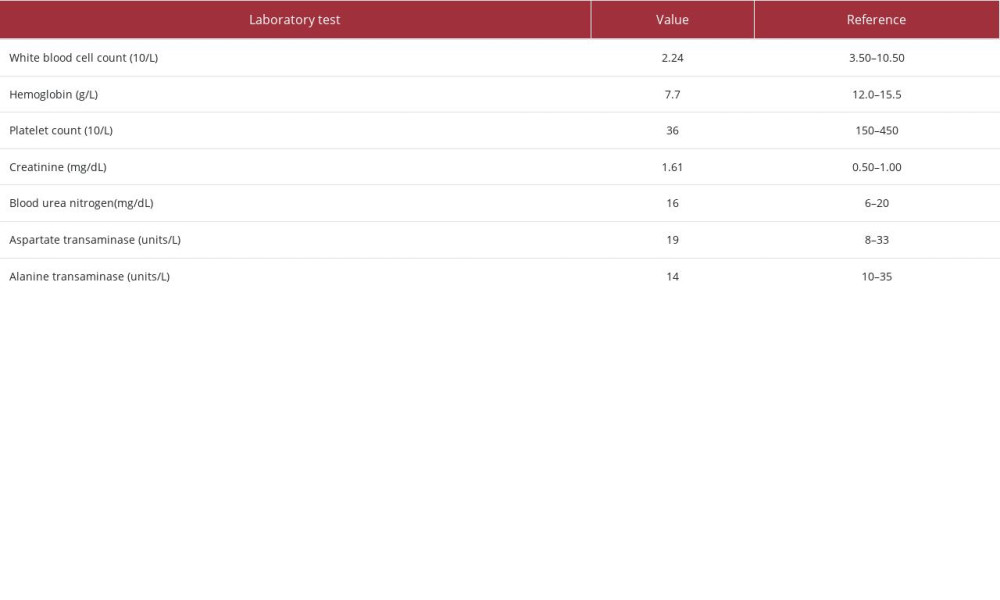 Table 1.. Summary of the pertinent initial laboratory findings of the patient.
Table 1.. Summary of the pertinent initial laboratory findings of the patient.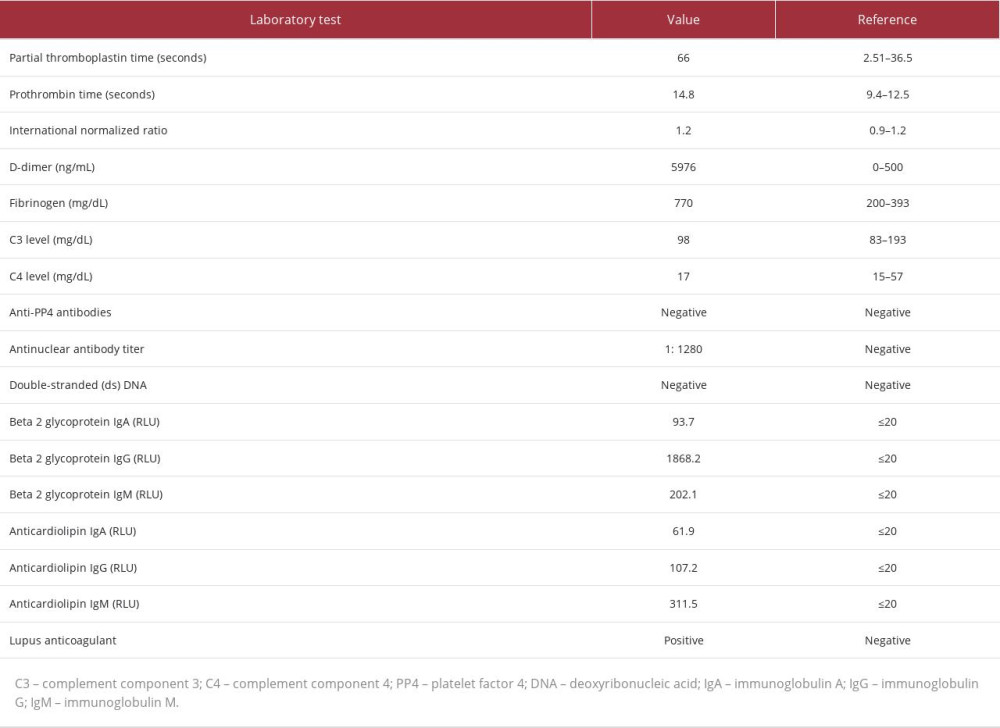 Table 2.. Summary of laboratory findings to further investigate etiology of thrombosis and thrombocytopenia.Table 1.. Summary of the pertinent initial laboratory findings of the patient.Table 2.. Summary of laboratory findings to further investigate etiology of thrombosis and thrombocytopenia.
Table 2.. Summary of laboratory findings to further investigate etiology of thrombosis and thrombocytopenia.Table 1.. Summary of the pertinent initial laboratory findings of the patient.Table 2.. Summary of laboratory findings to further investigate etiology of thrombosis and thrombocytopenia. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Most Viewed Current Articles
07 Dec 2021 : Case report
22,759,422
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  175,936
175,936
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,499
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,510
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133