07 April 2024: Articles 
Giant Bilateral Adrenal Myelolipomas in a Non-Compliant Patient with Congenital Adrenal Hyperplasia
Rare disease
Tomas Brutvan1BCDEF*, Otakar Psenicka1BC, Jarmila Krizova1BCD, Marcela Kotasova2BC, Jana Jezkova1ADEFDOI: 10.12659/AJCR.943005
Am J Case Rep 2024; 25:e943005






Abstract
BACKGROUND: 21-hydroxylase deficiency, an essential enzyme for glucocorticoid and mineralocorticoid synthesis, is the cause of congenital adrenal hyperplasia (CAH) in more than 95% of cases. It is an autosomal recessive disorder encoded by the CYP21A2 gene, categorized into classical forms, which encompass the salt-wasting (SW) and simple virilizing (SV) forms, as well as the nonclassical form (NC). The aim of medical treatment is to replace missing glucocorticoids and, if necessary, mineralocorticoids, while also reducing elevated adrenal androgens.
CASE REPORT: We present the case of a 42-year-old woman with CAH who discontinued therapy during adolescence and was admitted to hospital with fatigue, nausea, and severe abdominal pain. A CT scan showed an extreme enlargement of the adrenal glands. Laboratory tests revealed elevated levels of 17-hydroxyprogesterone and other adrenal androgens, along with normal plasma metanephrine levels. Decreased morning cortisol levels suggested partial adrenal insufficiency requiring glucocorticoid replacement therapy. Due to the development of several serious complications and clinical deterioration, the multidisciplinary team recommended bilateral removal of masses measuring 300×250×200 mm on the right side and 250×200×200 mm on the left side. Histological and immunochemical examination confirmed the presence of giant myelolipomas with adrenal cortex hyperplasia.
CONCLUSIONS: Adrenal tumors, particularly myelolipomas, have a higher prevalence in patients with CAH. Our case report provides further evidence of the suspected link between non-compliant CAH therapy and the development of myelolipomas, along with promotion of their pronounced growth.
Keywords: adrenal insufficiency, Adrenal incidentaloma, Congenital Adrenal Hyperplasia Due to 21 Hydroxylase Deficiency, Myelolipoma
Introduction
Congenital adrenal hyperplasia (CAH) is caused by an enzymatic disorder in adrenal steroidogenesis. In more than 95% of cases this disorder stems from 21-hydroxylase (21-OH) deficiency, which disrupts the conversion of 17-OH progesterone (17-OHP) into 11-deoxycortisol [1]. Other rare forms of CAH include 3β-hydroxysteroid dehydrogenase deficiency, P 450 oxidoreductase deficiency, and 11β-hydroxylase deficiency associated with mutations in the 3β-hydroxysteroid dehydrogenase (HSD3B2) and 11β-hydroxylase (CYP11B1) genes [2]. Based on the severity of 21-OH deficiency, a distinction is made between the classic form, with a prevalence of around 1: 15000 live births (based on infant screening), and the non-classic form (late onset), with a prevalence of around 1: 200 [3,4]. 21-OH deficiency is an autosomal recessive disorder encoded by the CYP21A2 gene. A large mutation in this gene results in no enzyme activity and causes the “salt-wasting” form of classic CAH (75 % of individuals) or the “simple virilizing” form in individuals with enzyme activity of around 1–2 % (25% of individuals). Individuals with enzyme activity around 5–20% have the non-classic (least severe) form of CAH [5].
Adult females present with clinical signs of androgen excess such as hirsutism, acne, menstrual irregularities, and infertility. In most cases, adult males are asymptomatic, and the diagnosis is often made based on family history.
The biochemical criteria for 21-OH deficiency are the same for men and women. Morning serum 17-OHP levels greater than 1000 ng/dl (30 nmol/l) confirm the diagnosis of CAH. Levels at 200–1000 ng/l (6–30 nmol/l) increase suspicion and should be confirmed by a high-dose adrenocorticotropic hormone (ACTH) stimulation test (250 ug), during which serum 17-OHP should rise above 1000 ng/l (30 nmol/l) to confirm the diagnosis [1,6].
The aim of therapy for CAH is to replace the missing steroids, suppress ACTH secretion, and reduce androgen production, with a dose that causes as few adverse effects as possible [1,7,8]. For the treatment of the classic form with salt wasting in adult patients, we use short- or long-acting glucocorticoids and mineralocorticoid substitution. For asymptomatic adults with non-classic congenital adrenal hyperplasia (NCCAH), therapy is generally not recommended. However, glucocorticoid therapy is suggested for women with NCCAH experiencing significant hyperandrogenism or infertility. Similarly, in men with NCCAH who face infertility, testicular adrenal rest tumors, or adrenal tumors, glucocorticoid therapy is also recommended [6].
Unilateral and bilateral adrenal hyperplasia or tumors have a higher incidence in patients with CAH than in the rest of the population. The most common are adrenocortical adenomas and myelolipomas. The etiology of their development is still unclear. It has been suggested that poor compliance with medication and high ACTH levels promote the growth of these tumors [9,10]. We present the case of a 42-year-old woman with CAH and massive bilateral adrenal tumors.
Case Report
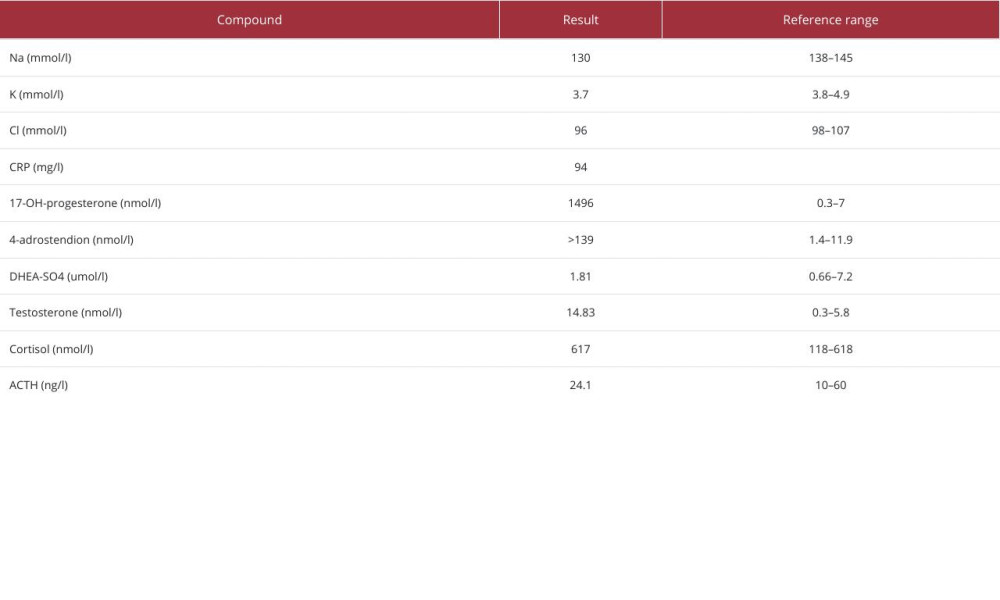
A 42-year-old woman was referred to the General University Hospital in Prague due to fatigue, nausea, vomiting, and severe pain in the epigastrium and the left side of the abdomen. On physical examination, the abdomen was above the air-fluid level (the niveau) and there was tenderness on palpation. A CT scan of the abdomen was performed at the request of the surgeon (Figure 1) and showed massive bilateral retroperitoneal tumors, measuring approximately 161×131×200 mm on the right side and 124×66×186 mm on the left side. The tumors were heterogeneous, with multiple cystic septa, but with clearly defined margins. In the region of the right ovary, other formations measuring 20×16 mm and 16×9 mm were also visible. The transverse colon, stomach, and pancreas were edematous, and ascitic fluid was present in the small pelvis. The left kidney was markedly reduced in volume and had become incorporated into the mass of the left tumor. In the differential diagnosis, the radiologist considered cystic pheochromocytoma, giant myelolipoma, or hemorrhage. Laboratory tests showed hypokalemia, hyponatremia, and elevated CRP (Table 1). The patient also exhibited signs of virilization: hirsutism and deepening of the voice.
The patient was referred to the 3rd Department of Internal Medicine, which has expertise in endocrine disorders.
In her medical history, the patient reported that she had been diagnosed with CAH and was treated with glucocorticoids, specifically hydrocortisone, in the past. She had undergone urogenital reconstruction surgery as a child. She discontinued hydrocortisone treatment a long time ago, so she was non-compliant and had a long history of socioeconomic hardship.
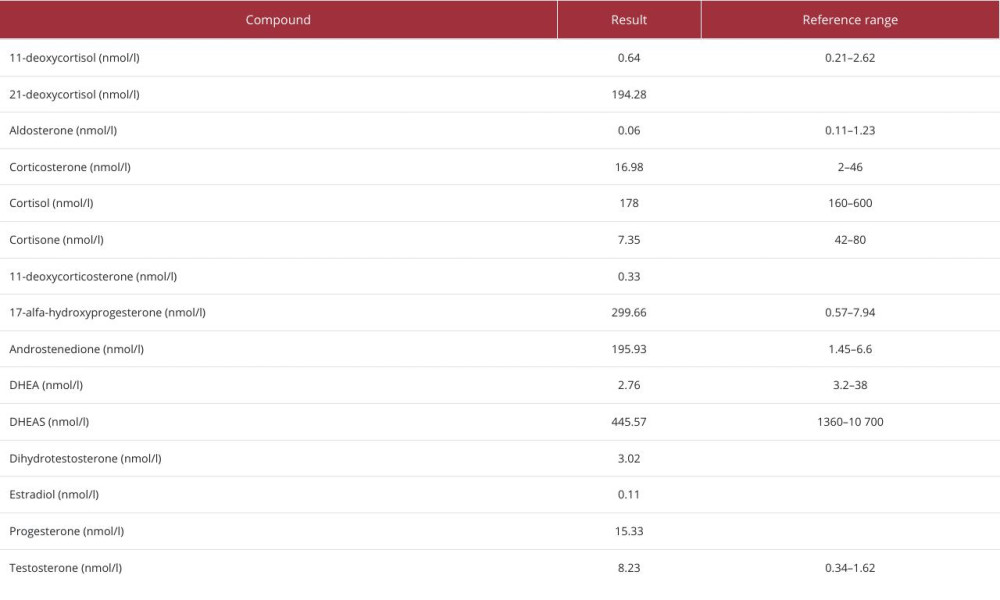
Laboratory tests using immunological methods revealed elevated levels of 17-OHP and testosterone, but she had normal morning cortisol levels (Table 1). Pheochromocytoma was ruled out by the normal plasma metanephrine levels. Two days after admission, she developed septic shock with multi-organ dysfunction (MODS) and was transferred to the Intensive Care Unit (ICU), requiring extensive fluid resuscitation, catecholamines, and antibiotics. Shortly after ICU admission, the results of laboratory analysis of steroid spectra by 2D-liquid chromatography-tandem mass spectrometry (2D-LC-MS/MS) were available, showing elevated adrenal steroid levels and plasma cortisol levels at the lower limit of the reference interval (Table 2). Due to the high likelihood of partial adrenal insufficiency, glucocorticoid replacement therapy was initiated at doses consistent with the treatment of acute adrenal crisis. Her renal function deteriorated and intra-abdominal pressure (IAP) reached about 18 mm of water column. The patient developed abdominal compartment syndrome and oliguria with a diuresis of less than 100 ml per 24 h. We started continuous renal replacement therapy (CRRT) and, due to the rapid clinical deterioration, the case was presented to a multidisciplinary surgical team. She was referred for bilateral mass removal and the operation was performed without any acute complications.
A tumor measuring300×250×200 mm was removed from the right side and another measuring 250×200×200 mm from the left side. The atrophic left kidney was also removed. The histology of both masses showed the characteristics of a myelolipoma, with the predominance of edematous altered adipose tissue, hematopoietic elements and, mainly in the periphery, signs of an enlarged adrenal cortex of variable thickness with the predominance of zona reticularis cells. The presence of the adrenal cortex was confirmed by immunohistochemistry- positive Inhibin and Melan A antibodies (Figure 2).
As removal of the masses meant bilateral adrenalectomy, we started glucocorticoid and mineralocorticoid replacement therapy. The patient’s recovery was successful, but 3 weeks after the operation, the patient decided to end her hospitalization, despite medical advice to stay in the hospital and continue her recovery, as physiotherapy, nutrition supervision, and antibiotic therapy were still needed. The patient was discharged with a prescription for glucocorticoid and mineralocorticoid replacement therapy and was instructed to take the medication regularly, otherwise her life could be threatened.
After discharge, the patient was readmitted to the hospital twice for signs of hypocortical crisis and died 11 weeks after surgery.
The autopsy was inconclusive as to the cause of death, but it was most likely due to an untreated hypocortical crisis.
Discussion
We present the case of a 42-year-old woman with CAH who was non-compliant with therapy and subsequently was admitted to the hospital with abdominal pain and gigantic bilateral retroperitoneal tumors.
Adrenal incidentalomas are mass lesions larger than 1 cm in diameter that are discovered on imaging performed for a reason other than suspected adrenal disease. The prevalence of adrenal incidentalomas on abdominal CT is approximately 2–4% and increases with age. In 10–15% of cases, incidentalomas are bilateral [11].
The etiology of bilateral adrenal masses is varied and includes bilateral benign or malignant tumors (functional or non-functional), bilateral macronodular adrenal hyperplasia, metastatic lesions, tuberculosis, amyloidosis, and hemorrhage. Individual lesions are characterized by specific features on CT or MRI scans.
Benign adenomas tend to have a round shape and homogeneous density, a smooth contour with a border, a sharp margin, and a density on non-contrast CT ≤10 HU. Pheochromocytomas, on the other hand, display a higher density on non-contrast CT (>20 HU), increased vascularity of the mass, cystic or hemorrhagic changes, and variable size, and can be bilateral. In contrast, adrenal carcinomas are typically larger than 4 cm in diameter, heterogeneous, irregular, unilateral, often present with metastases or local invasion, and exhibit high density on noncontrast CT (>20 HU). Metastases also tend to be irregular with high density, but are commonly bilateral [12]. Imaging followup of adrenal incidentalomas is based on a feature on unenhanced CT, which is outlined in the clinical practice guidelines of the European Society of Endocrinology [13].
As the adrenal masses were heterogeneous on the CT scan and gigantic in size (larger than 4 cm), in accordance with European Society of Endocrinology practice guidelines, we discussed the case in a multidisciplinary team [13]. Non-elevated plasma metanephrine levels excluded the radiological suspicion of pheochromocytoma. Primary tumor or metastasis were less probable, as the tumors were bilateral, had distinct margins, and there were no calcifications, other metastatic lesions, or lymphadenopathy.
Surgery, specifically bilateral adrenalectomy, was indicated due to the development of complications, including abdominal compartment syndrome, suspicion of intratumoral hemorrhage, and the unclear biological nature of the lesion.
Histologically, the tumors were identified as myelolipomas with adrenal cortex hyperplasia, which was confirmed by immunohistochemistry. Myelolipomas are recognized as benign neoplasms of the adrenal cortex, consisting of fatty and myeloid tissue. Their incidence is 0.08–0.4 % and represents 3.3–6.5% of all adrenal masses. Most myelolipomas are unilateral, with a median tumor size of 2–2.5 cm. Bilateral tumors tend to be larger (>6 cm) and are often associated with abdominal discomfort. The precise pathogenesis of myelolipomas is still unclear. One hypothesis suggests that the primary event involves a metaplastic change due to necrosis or infection in the reticuloendothelial cells of the blood capillaries. It has also been speculated that adrenal myelolipoma is derived from embryonic primitive mesenchymal cells [14].
Another hypothesis, related to hormonal influence and based on clinical observation, posits that some patients with untreated CAH who exhibit high serum adrenocorticotropin (ACTH) levels develop myelolipoma. Meta-analyses have shown that 23–28% of patients with 21-OH deficiency have developed an adrenal tumor, and, based on published case reports, almost all patients with late diagnosed CAH or poorly treated disease have myelolipoma [15–17]. Steroid receptors in myelolipomas have been studied in 2 published articles. Almeida et al demonstrated overexpression of melanocortin 2 receptor (MCR2) gene in 3 out of 4 myelolipomas. Androgen receptor (AR) over-expression was detected in 2 myelolipomas. The overexpression of AR and MC2R genes was concomitantly found in 2 of 4 cases. In contrast, low levels of MC2R, MC5R or AR gene expression were only found in a single case of giant myelolipoma tissue by Hagiwara et al. The observed overexpression of MC2R or AR suggest that chronically increased levels of ACTH and androgens are involved in the pathogenesis of myelolipomas. Furthermore, it is plausible that these receptors are differentially expressed at various stages of tumor development, possibly more prominently in the early stages [18,19]. However, the precise role of hormonal stimulation in the pathogenesis of myelolipomas remains unclear and needs further studies.
In most cases, myelolipomas do not cause symptoms because they are mostly hormonally inactive and small. If symptoms do occur (usually abdominal pain), surgical removal is the preferred approach. Some surgeons suggest that asymptomatic giant myelolipomas should be removed as well, as the risk of rupture or bleeding tends to increase with the tumor’s size. Nevertheless, there is no definitive consensus regarding the best surgical approach for myelolipomas and what size of myelolipoma is an indication for surgical removal [15,20,21].
Cross-reactions of steroids analyzed by immunoassay are a common pitfall of this laboratory method [22,23]. If CAH is suspected, the European Society of Endocrinology clinical practice guideline recommends steroid analysis by LC-MS/MS [6]. The significantly different cortisol levels found by immunomethod and 2D-LC-MS/MS are explained by the cross-reactivity of endogenous steroids immunomethod measurements. Untreated or poorly compensated CAH is associated with elevated androgen levels, which are involved in this cross-reactivity. Elevated levels of androgens, including 17-OHP and 21-deoxycortisol, were indicative of 21-hydroxylase deficiency. Janzen et al proposed calculation of the ratio (21-deoxycortisol+17-OHP)/cortisol analyzed by LC-MS/MS to detect 21-hydroxylase deficiency. A ratio of 0.516–12.7 supports this diagnosis, with specificity, sensitivity, and a positive predictive value of 1.0. In our case, the patient’s ratio was 2.77 [24], but genetic testing was not performed. The results of hormonal analysis and the patient’s condition following the genitourinary surgery in her childhood are indicative of the classic “simple virilizing” form of CAH.
The structure seen on the CT scan near the right ovary could be considered an adrenal residual tumor (ART), a known complication associated with poorly controlled CAH. ARTs can develop in many locations. For example, testicular adrenal tumors (TARTs) are very typical for male patients with CAH, while female patients can develop ovarian adrenal tumors (OARTs). ARTs can also occur in less typical sites, such as the lungs and pituitary gland [9].
After bilateral adrenalectomy, lifelong replacement therapy with glucocorticoids and mineralocorticoids becomes necessary. It is recommended to use hydrocortisone (15–25 mg) or cortisone acetate (20–35 mg) in 2 or 3 divided oral doses per day. Hydrocortisone is available in the Czech Republic. As an alternative to hydrocortisone, prednisolone (3–5 mg/d) administered orally once or twice a day is recommended, especially in patients with reduced compliance. Each patient must be aware of the signs indicating an impending crisis and the need to increase the replacement dose of glucocorticoids in stressful situations [25]. The annual incidence of adrenal crisis ranges is 5.2–8.3% in patients with adrenocortical insufficiency, and, if untreated, will result in death [26].
Conclusions
Our case report provides further confirmation that patients with CAH, particularly those with poor compliance, face a higher risk of developing adrenal myelolipomas, along with their pronounced growth promotion [16,18].
Figures
References:
1.. Merke DP, Auchus RJ, Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: N Engl J Med, 2020; 383(13); 1248-61
2.. Witchel SF, Congenital adrenal hyperplasia: J Pediatr Adolesc Gynecol, 2017; 30(5); 520-34
3.. Therrell BL, Newborn screening for congenital adrenal hyperplasia: Endocrinol Metab Clin North Am, 2001; 30(1); 15-30
4.. Hannah-Shmouni F, Morissette R, Sinaii N, Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in US Ashkenazi Jews and Caucasians: Genet Med, 2017; 19(11); 1276-79
5.. Speiser PW, Dupont J, Zhu D, Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency: J Clin Invest, 1992; 90(2); 584-95
6.. Speiser PW, Arlt W, Auchus RJ, Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An endocrine society clinical practice guideline: J Clin Endocrinol Metab, 2018; 103(11); 4043-88
7.. Merke DP, Poppas DP, Management of adolescents with congenital adrenal hyperplasia: Lancet Diabetes Endocrinol, 2013; 1(4); 341-52
8.. Whitaker M, Debono M, Huatan H, An oral multiparticulate, modified-release, hydrocortisone replacement therapy that provides physiological cortisol exposure: Clin Endocrinol (Oxf), 2014; 80(4); 554-61
9.. Kolli V, da Cunha IW, Kim S, Morphologic and molecular characterization of adrenals and adrenal rest affected by congenital adrenal hyperplasia: Front Endocrinol (Lausanne), 2021; 12; 730947
10.. El-Maouche D, Hannah-Shmouni F, Mallappa A, Adrenal morphology and associated comorbidities in congenital adrenal hyperplasia: Clin Endocrinol (Oxf), 2019; 91(2); 247-55
11.. Arnaldi G, Boscaro M, Adrenal incidentaloma: Best Pract Res Clin Endocrinol Metab, 2012; 26(4); 405-19
12.. Terzolo M, Stigliano A, Chiodini I, Italian Association of Clinical Endocrinologists: AME position statement on adrenal incidentaloma. Eur J Endocrinol, 2011; 164(6); 851-70
13.. Fassnacht M, Tsagarakis S, Terzolo M, European Society of Endocrinology clinical practice guidelines on the management of adrenal incidentalomas, in collaboration with the European Network for the Study of Adrenal Tumors: Eur J Endocrinol, 2023; 189(1); G1-G42
14.. Feng C, Jiang H, Ding Q, Wen H, Adrenal myelolipoma: A mingle of progenitor cells?: Med Hypotheses, 2013; 80(6); 819-22
15.. Al-Bahri S, Tariq A, Lowentritt B, Nasrallah DV, Giant bilateral adrenal myelolipoma with congenital adrenal hyperplasia: Case Rep Surg, 2014; 2014; 728198
16.. Nermoen I, Falhammar H, Prevalence and characteristics of adrenal tumors and myelolipomas in congenital adrenal hyperplasia: A systematic review and meta-analysis: Endocr Pract, 2020; 26(11); 1351-65
17.. Wang J, Bissada MA, Williamson HO, Adrenal tumors associated with inadequately treated congenital adrenal hyperplasia: Can J Urol, 2002; 9(3); 1563-64
18.. Almeida MQ, Kaupert LC, Brito LP, Increased expression of ACTH (MC2R) and androgen (AR) receptors in giant bilateral myelolipomas from patients with congenital adrenal hyperplasia: BMC Endocr Disord, 2014; 14; 42
19.. Hagiwara H, Usui T, Kimura T, Lack of ACTH and androgen receptor expression in a giant adrenal myelolipoma associated with 21-hydroxylase deficiency: Endocr Pathol, 2008; 19(2); 122-27
20.. Decmann Á, Perge P, Tóth M, Igaz P, Adrenal myelolipoma: A comprehensive review: Endocrine, 2018; 59(1); 7-15
21.. Calissendorff J, Juhlin CC, Sundin A, Adrenal myelolipomas: Lancet Diabetes Endocrinol, 2021; 9(11); 767-75
22.. Brossaud J, Barat P, Gualde D, Corcuff JB, Cross reactions elicited by serum 17-OH progesterone and 11-desoxycortisol in cortisol assays: Clin Chim Acta, 2009; 407(1–2); 72-74
23.. Krasowski MD, Drees D, Morris CS, Cross-reactivity of steroid hormone immunoassays: Clinical significance and two-dimensional molecular similarity prediction: BMC Clin Pathol, 2014; 14; 33
24.. Janzen N, Peter M, Sander S, Newborn screening for congenital adrenal hyperplasia: Additional steroid profile using liquid chromatography-tandem mass spectrometry: J Clin Endocrinol Metab, 2007; 92(7); 2581-89
25.. Bornstein SR, Allolio B, Arlt W, Diagnosis and treatment of primary adrenal insufficiency: An Endocrine Society clinical practice guideline: J Clin Endocrinol Metab, 2016; 101(2); 364-89
26.. Dineen R, Thompson CJ, Sherlock M, Adrenal crisis: Prevention and management in adult patients: Ther Adv Endocrinol Metab, 2019; 10; 2042018819848218
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133