18 April 2024: Articles ")
Transformation of Severe Aplastic Anemia into Donor Cell Leukemia after Allogeneic Hematopoietic Stem Cell Transplantation: A Rare Case Report
Rare disease
Qianqian Wang1ABCDEFG, Hong Xu1ABCD, Wei Yu1BC, Lingjie Sun1BC, Hongguo Zhao1BC, Xue Shi1ADF*DOI: 10.12659/AJCR.943801
Am J Case Rep 2024; 25:e943801






Abstract
BACKGROUND: Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is an important treatment for severe aplastic anemia (SAA). It is known that SAA can evolve into malignant clonal diseases, such as acute myeloblastic leukemia (AML) or myelodysplastic syndrome. However, the transformation of SAA into AML after allo-HSCT is a rare phenomenon. Here, we report a case of SAA transformed into AML after patient received human leucocyte antigen (HLA)-matched sibling peripheral blood stem cell transplantation.
CASE REPORT: A 51-year-old female patient presented with petechiae and fatigue and received a diagnosis of idiopathic SAA. The immunosuppressive therapy combined with umbilical cord blood transplantation failed for this patient. Then, she received HLA-matched sibling allogeneic peripheral blood stem cell transplantation (allo-PBSCT). However, 445 days after allo-PBSCT, the patient had a diagnosis of AML by bone marrow puncture. Donor-recipient chimerism monitoring and cytogenetic analysis confirmed that the leukemia was donor cell origin. Notably, a new HOXA11 mutation was detected in the peripheral blood of the patient after transplantation by whole-exome sequencing, which was the same gene mutation detected in the donor. The patient received 1 cycle of induction chemotherapy with azacytidine and achieved complete remission. However, the leukemia relapsed after 2 cycles of consolidation chemotherapy. Unfortunately, the patient died of leukemia progression 575 days after allo-HSCT.
CONCLUSIONS: The mechanism of how normal donor hematopoietic cells transform to leukemia in the host remains unclear. Donor cell leukemia provides a unique opportunity to examine genetic variations in donors and hosts with regards to the progression to malignancy.
Keywords: Hematopoietic Stem Cell Transplantation, Leukemia, Aplastic Anemia, Idiopathic, Female, Humans, Middle Aged, Anemia, Aplastic, Graft vs Host Disease, Tissue Donors, Leukemia, Myeloid, Acute, HLA Antigens
Introduction
Aplastic anemia (AA) is an immune-mediated bone marrow failure disease. Previous studies have reported that AA can evolve into malignant clonal diseases, such as myelodysplastic syndrome (MDS) or acute myeloblastic leukemia (AML), with a transformation rate of 10% to 20% [1]. Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the recommended treatment for patients with severe aplastic anemia (SAA) who have suitable donors and are younger than 40 years old or have failed in immunosuppressive therapy, including anti-thymocyte globulin (ATG) and cyclosporine A [2]. The development of secondary malignancies is one of the long-term complications after transplantation. Donor cell leukemia (DCL) is defined as leukemia that develops in engrafted cells of donor origin. Studies have found that DCL can account for more than 2% of leukemia relapses after transplantation [3]. However, only 5 cases of DCL transformed from SAA after allo-HSCT have been reported to date [4–8]. With the development of whole-exome and genome sequencing, some candidate genes that may contribute to the occurrence of DCL have been detected [3]. Here, we present a case of DCL transformed from SAA 445 days after human leucocyte antigen (HLA)-matched sibling peripheral blood stem cell transplantation. In addition, we found a new
Case Report
A 51-year-old female patient was admitted to our hospital with petechiae and fatigue in April 2021. The complete blood count (CBC) showed pancytopenia, with a white blood cell count of 2.13×109 cells/L, hemoglobin level of 79 g/L, and platelet count of 25×109 cells/L. The neutrophil count was 0.38×109 cells/L, and the reticulocyte count was 0.02×1012 cells/L. Tests for viral infections, including hepatitis virus, HIV, syphilis, cytomegalo-virus, and Epstein-Barr virus, were negative. Autoimmune diseases were excluded by serological tests. There was no indication of hemolytic anemia, nutritional anemia, or paroxysmal nocturnal hemoglobinuria. Bone marrow smear showed marked hypoplasia and contained mainly lymphocytes (Figure 1A, 1B). Bone marrow biopsy showed about 10% of cellularity, and most of the hematopoietic tissue was replaced by adipose tissue (Figure 1C, 1D). Furthermore, no megakaryocytes or blast cells were identified on bone marrow smears or biopsy specimens. Acute leukemia and MDS were further excluded by immunophenotype analysis. Conventional cytogenetic analysis showed a normal 46, XX karyotype in 24 metaphases. Mitomycin C induced peripheral blood chromosome breakage detection, and chromosome aberration tests were normal. Comet assay was used to detect the DNA damage in peripheral blood lymphocytes. Comet cells were formed when fragmented DNA migrated out of the nucleoid (also known as the “comet head”) and formed a DNA stain in the agarose gel (also known as the “comet tail”). The percentage of comet cells was 16%. There was no history of exposure to drugs, chemicals, or radiation. The patient’s family members were all healthy and none of them had similar blood disorders. There was no history of consanguineous marriage in her family. The patient had no symptoms of infection. Physical examination revealed pallor and petechiae. The patient did not have any developmental abnormalities. Therefore, idiopathic SAA was diagnosed.
The patient received immunosuppressive therapy combined with umbilical cord blood transplantation in April 2021. A total of 31.8 mL autologous frozen umbilical cord full blood was infused. The immunosuppressive therapy regimen consisted of rabbit ATG (3.5 mg/kg/d×5 days, intravenously) and cyclosporine A (3-5 mg/kg, orally, dosage adjusted according to drug concentration). The patient was treated with granulocyte colony stimulating factor (G-CSF) for 14 days. The granulocytes were engrafted at day 14 (neutrophils >0.5×109 cells/L for 3 consecutive days). Donor-recipient chimerism analysis by short tandem repeat was 96.97% at day 36. However, the hemoglobin level and the platelet count did not return to normal. Bone marrow smear and biopsy at day 104 showed marked hypoplasia, and no megakaryocytes were seen. The immunosuppressive therapy combined with umbilical cord blood transplantation failed for this patient.
In November 2021, the diagnosis of idiopathic SAA was confirmed by a bone marrow puncture performed at People’s Hospital of Peking University. The patient received HLA-matched sibling allogeneic peripheral blood stem cell transplantation (allo-PBSCT). The donor was her 49-year-old younger brother. Before transplantation, the donor had comprehensive examinations, including CBC, coagulation routine test, liver and kidney function, viral infection tests, urinalysis, stool routine test, electrocardiogram, echocardiography, chest X-ray, and abdominal ultrasound. All these examinations were normal. The results of bone marrow morphology, biopsy, immunophenotype analysis, and chromosome karyotype analysis confirmed that the hematopoietic function of the donor was normal. Psychological assessment was performed to exclude psychological disorders, such as schizophrenia and depression. Physical examination of the donor was normal. The donor had no vascular complications or other diseases. The donor was demonstrated to be clinically healthy.
Before allo-PBSCT, whole-exome sequencing was performed on the patient to further exclude congenital bone marrow failure. Only a heterozygous missense mutation was detected in
However, 445 days after allo-PBSCT, the CBC showed anemia and thrombocytopenia with leukocytosis. The white blood cell count was 65.79×109 cells/L, hemoglobin level was 71 g/L, and platelet count was 10×109 cells/L. Bone marrow smear and biopsy were consistent with the diagnosis of AML (Figure 2A, 2B). The immunophenotype analysis showed that 45.2% of the blast cells had monocytic features (AML-M5). The conventional cytogenetic analysis showed a normal donor karyotype of 46, XY in 3 metaphases. Short tandem repeat remained complete donor chimerism.
Discussion
The mechanism of how normal donor hematopoietic cells transform to leukemia in the host remains unclear. Hypotheses includes occult leukemia or preleukemic potential in the donor, leukemic transformation of engrafted cells, transfer of oncogenic material from host cells to donor cells, residual effects of cytotoxic chemotherapy or radiotherapy, defective marrow microenvironment, inadequate immune surveillance result from post-transplant immunosuppression, viral transfection/integration, and telomere shortening and replicative stress [9]. In addition, prolonged use of G-CSF has been reported as a risk factor for the malignant evolution of SAA [10]. However, G-CSF use is usually associated with poor granulocytes reconstruction after immunosuppressive therapy or transplantation; therefore, the impact of G-CSF cannot be accurately assessed. Monosomy 7 arises as a recurrent chromosome aberration in DCL [6] and has been previously described in DCL after bone marrow transplantation for SAA. In the setting of decreased hematopoietic stem progenitor cells pools, accelerated telomere attrition is considered a possible mechanism for early myeloid oncogenesis and aneuploidy development in MDS/AML after SAA [10].
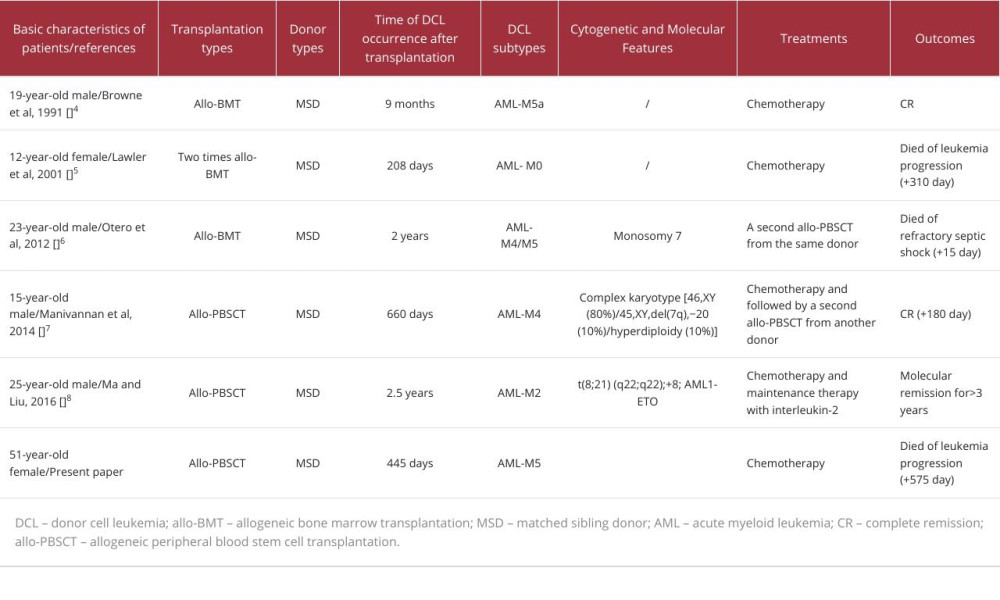
DCL is a rare but severe post-transplant complication. Most cases of DCL are primary AML, acute lymphoblastic leukemia, or MDS, accounting for 93% of all reported cases [11]. Only 5 cases of DCL transformed from SAA after allo-HSCT have been reported. Generally, the prognosis of DCL is poor, and there is no recommended standard treatment strategy at present. Standard AML- or acute lymphoblastic leukemia-specific regimens can induce complete remission in DCL; however, the treatment-related mortality is high. Considering the graft-vs-leukemia effect, secondary hematopoietic stem cell transplantation from an alternative donor remains a more logical treatment strategy [9]. For DCL transformed from SAA, treatment experience was obtained from case report studies. Among the 5 reported cases, 1 patient died of refractory septic shock after receiving a second allo-HSCT from the same donor [6]. Another 1 patient died of leukemia progression after receiving intensive chemotherapy consisting of idarubicin (10 mg/m2/day for 3 days) and cytarabine (300 mg/m2/day for 7 days) [5].
The other 3 patients are alive. One patient was treated with standard 3+7 induction chemotherapy with cytarabine and daunorubicin and then received a second allo-HSCT from an alternative donor [7]. One patient obtained remission with chemotherapy [4]. Another patient remained molecular remission for more than 3 years with chemotherapy and IL-2 maintenance therapy [8]. A brief comparison of the similarities and differences between our case and the other 5 cases is summarized in Table 1.
In our case, the patient received immunosuppressive therapy combined with umbilical cord blood transplantation and allo-PBSCT successively, leading to long-term immunosuppression and impaired immune surveillance, which may have contributed to the development of leukemia. The G-CSF was not used for a long time, and no chromosomal aberrations were found in this patient.
Clonal hematopoiesis is a strong risk factor for subsequent hematologic cancer. Donor clonal hematopoiesis is closely related to the clinical outcomes of transplant recipients, with differential impact on graft alloimmune function and potential for leukemic transformation related to mutated gene and somatic clonal abundance [19].
There are some other limitations in our study. First, due to the scattered and heterogeneous nature of cases, the mechanisms and treatment strategies of DCL transformed from SAA need to be further explored. Second, there was a lack of functional experiment to elucidate the function of
Conclusions
In conclusion, the precise mechanism of SAA transformed into DCL after allo-HSCT remains unclear. In the present case, the unique status of bone marrow microenvironment may have played a crucial role in the development of DCL. Leukemogenesis is more than a 1-step process, and DCL provides a unique opportunity to examine genetic variations in donors and hosts with regards to the progression to malignancy.
Figures
References:
1.. Bakanay ŞM, Topçuoğlu P, Uğur Bilgin A, Clonal evolution of monosomy 7 in acquired severe aplastic anemia: Two cases treated with allogeneic hematopoietic stem cell transplantation: Turk J Haematol, 2008; 25(2); 94-97
2.. Killick SB, Bown N, Cavenagh J, Guidelines for the diagnosis and management of adult aplastic anaemia: Br J Haematol, 2016; 172(2); 187-207
3.. Williams L, Doucette K, Karp JE, Genetics of donor cell leukemia in acute myelogenous leukemia and myelodysplastic syndrome: Bone Marrow Transplant, 2021; 56(7); 1535-49
4.. Browne PV, Lawler M, Humphries P, Donor-cell leukemia after bone marrow transplantation for severe aplastic anemia: N Engl J Med, 1991; 325(10); 710-13
5.. Lawler M, Locasciulli A, Longoni D, Leukaemic transformation of donor cells in a patient receiving a second allogeneic bone marrow transplant for severe aplastic anaemia: Bone Marrow Transplant, 2002; 29(5); 453-56
6.. Otero L, de Souza DC, de Cássia Tavares R, Monosomy 7 in donor cell-derived leukemia after bone marrow transplantation for severe aplastic anemia: Report of a new case and review of the literature: Genet Mol Biol, 2012; 35(4); 734-36
7.. Manivannan P, Purohit A, Somasundaram V, Leukemic transformation of severe aplastic anemia following matched allogenic stem cell transplantation, transplanted again in CR 1: Indian J Hematol Blood Transfus, 2016; 32(Suppl 1); 223-27
8.. Ma H, Liu T, Development of donor cell leukemia following peripheral blood stem cell transplantation for severe aplastic anemia: A case report: Oncol Lett, 2016; 11(6); 3858-62
9.. Wiseman DH, Donor cell leukemia: A review: Biol Blood Marrow Transplant, 2011; 17(6); 771-89
10.. Dumitriu B, Feng X, Townsley DM, Telomere attrition and candidate gene mutations preceding monosomy 7 in aplastic anemia: Blood, 2015; 125(4); 706-9
11.. Suárez-González J, Martínez-Laperche C, Kwon M, Donor cell-derived hematologic neoplasms after hematopoietic stem cell transplantation: A systematic review: Biol Blood Marrow Transplant, 2018; 24(7); 1505-13
12.. Kam MLW, Nguyen TTT, Ngeow JYY, Telomere biology disorders: NPJ Genom Med, 2021; 6(1); 36
13.. Yang TP, Stout JT, Konecki DS, Spontaneous reversion of novel Lesch-Nyhan mutation by HPRT gene rearrangement: Somat Cell Mol Genet, 1988; 14(3); 293-303
14.. Wada T, Candotti F, Somatic mosaicism in primary immune deficiencies: Curr Opin Allergy Clin Immunol, 2008; 8(6); 510-14
15.. Zhang N, Miao XJ, Shuai YR, Family aggregation of hematological malignancies discovered from an acute myeloid leukemia patient with STK11 and THBD gene mutation: Case Rep Oncol, 2023; 16(1); 734-38
16.. Mizoguchi Y, Fujita N, Taki T, Juvenile myelomonocytic leukemia with t(7;11)(p15;p15) and NUP98-HOXA11 fusion: Am J Hematol, 2009; 84(5); 295-97
17.. Li Z, Huang H, Li Y, Up-regulation of a HOXA-PBX3 homeobox-gene signature following down-regulation of miR-181 is associated with adverse prognosis in patients with cytogenetically abnormal AML: Blood, 2012; 119(10); 2314-24
18.. Fu JF, Shih LY, Yen TH, HOXA11 plays critical roles in disease progression and response to cytarabine in AML: Oncol Rep, 2021; 46(1); 150
19.. Gibson CJ, Kim HT, Zhao L, Donor clonal hematopoiesis and recipient outcomes after transplantation: J Clin Oncol, 2022; 40(2); 189-201
20.. Bick AG, Weinstock JS, Nandakumar SK, Inherited causes of clonal haematopoiesis in 97,691 whole genomes: Nature, 2020; 586(7831); 763-68
Figures
In Press
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Most Viewed Current Articles
07 Dec 2021 : Case report  22,759,422
22,759,422
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  175,936
175,936
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,499
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,510
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133