13 July 2020: Articles 
Desmoplastic Small Round Cell Tumor of Pancreatic Origin in a Young Child: A Case Report and Review of Literature
Challenging differential diagnosis, Management of emergency care, Patient complains / malpractice, Rare disease, Adverse events of drug therapy, Clinical situation which can not be reproduced for ethical reasons
Daniyah Saleh1ABCDEFG, Sahar Al-Maghrabi2ABCDEFG, Haneen Al-Maghrabi1ABCDEFG, Jaudah Al-Maghrabi13ABCEFG*DOI: 10.12659/AJCR.922762
Am J Case Rep 2020; 21:e922762






Abstract
BACKGROUND: Desmoplastic small round cell tumor (DSRCT) is a rare lethal malignant tumor with young male predominance. The majority of cases arise in the abdominopelvic region and are hypothesized to have a mesothelial origin. However, extra-abdominal and extraperitoneal DSRCT have been reported. It is extremely uncommon for the pancreas to be a primary site for DSRCT, and only 5 cases have previously been reported in the English literature. Clinically, DSRCT has a wide range of presentations from asymptomatic to life-threatening comorbidity, and it responds poorly to treatment despite aggressive therapy.
CASE REPORT: We report a previously healthy 9-year-old boy with an incidentally discovered abdominal mass of pancreatic origin. All necessary laboratory investigations were within normal limits. Computed tomographic imaging showed a huge left-side retroperitoneal mass measuring 15 cm in the greatest dimension that was accompanied by vascular encasement. The mass was resected successfully. Histopathological examination along with ancillary tests favored a diagnosis of DSRCT over other small round blue cell tumors. Detection of translocation t(11;22)(p13;q12) with EWSR1-WT1 gene fusion, based on reverse transcription-polymerase chain reaction analysis, confirmed the diagnosis. Approximately 7 months later, the tumor recurred with mesenteric lymph nodes metastasis and the child was placed on palliative therapy.
CONCLUSIONS: It is worthwhile to consider DSRCT in the differential diagnosis of small round blue cell tumors, even in unusual sites, in a pediatric age group. Due to the poor prognosis, owing to chemotherapy resistance and a high rate of recurrence with significant tumor burden, reaching a precise diagnosis of DSRCT is essential. Almost all cases harbor the hallmark molecular alteration of t(11;22)(p13;q12) with EWSR1-WT1 gene fusion. Debulking surgery paired with a chemotherapy regimen comprising vincristine, doxorubicin, and cyclophosphamide and ifosfamide + etoposide has been shown to improve overall survival rate compared with other chemotherapeutic agents. However, no targeted therapeutic modality has been developed.
Keywords: desmoplastic small round cell tumor, Pancrelipase, Pediatrics, Child, Incidental Findings, Oncogene Proteins, Fusion, Pancreatic Neoplasms, RNA-Binding Protein EWS, Translocation, Genetic
Background
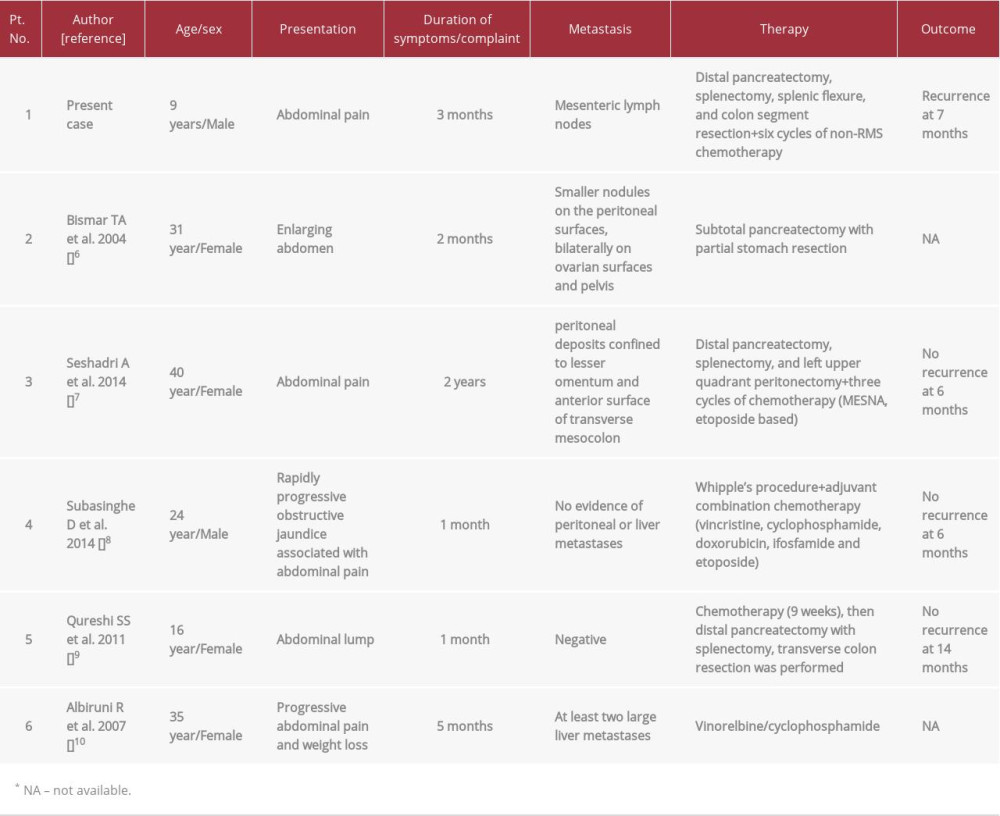
In 1989, desmoplastic small round cell tumor (DSRCT) was recognized as a separate entity by Gerald and Rosai [1]. This rare malignant tumor predominantly affects children and adolescent males, with a peak incidence in the third decade of life [2]. Usually, it arises from serosal surfaces, mainly the peritoneum. The second most common site is the abdominopelvic region, with no distinct organ involvement. However, DSRCT arising from various body organs, such as pleura, ovary, testis, central nervous system, orbit, and others, has been described [3,4]. Primary pancreatic DSRCT is extremely rare, limited only to 5 reported cases in the available English literature (Table 1). Since DSRCT has nonspecific imaging features, microscopic examination is very helpful because it shows malignant cells with primitive morphology of undifferentiated round blue cells separated by the cellular desmoplastic stromal reaction. In addition, immunohistochemistry expression of mixed mesenchymal, epithelial, and neural markers is exclusively supportive. The most accurate, well-established molecular analysis is by reverse transcription-polymerase chain reaction (RT-PCR) or fluorescence
Case Report
A previously healthy 9-year-old boy presented to a pediatric surgery clinic with a history of abdominal mass accompanied by intermittent abdominal pain during the 3 months before admission. The mass was incidentally discovered at an outside facility after the boy experienced a mild abdominal trauma. Physical examination demonstrated firmness on the left side of the abdomen. Otherwise, the child was alert and in good general condition. Laboratory investigations, including blood profile, renal and liver function tests, coagulation profile, electrolytes, and urine analysis, were within normal limits. Amylase was 165 U/L and lipase was 259 U/L; tumor markers were not performed. Abdominal computed tomography (CT) scan with contrast showed a left flank soft tissue abdominal mass (15×10.1×6.5 cm) that extended from the splenic hilum down to the level of the aorta bifurcation. The mass was associated with significant vascular encasement, and it was not separable from the pancreatic body and tail. No definite hepatic or splenic lesions were present (Figure 1A). Radiological impression included neuroblastoma, lymphoma, and neuroendocrine tumor. The patient underwent exploratory laparotomy for debulking and removal of the tumor with distal pancreatectomy, splenectomy, splenic flexure, and colon segment resection. Microscopic examination depicted an infiltrative neoplastic growth arranged in well-defined, relatively sharply demarcated nests embedded in the dense desmoplastic stroma. The neoplastic cells exhibited features of small round blue cells with increased nuclear to stromal ratio, hyperchromatic nuclei, and scarce cytoplasm (Figure 1B, 1C). To reach a specific diagnostic entity, a panel of immunohistochemistry markers were assayed. Neoplastic cells were immunoreactive to vimentin, desmin (Figure 1D), CD56 (Figure 1E), CD57, and neuron-specific enolase (Figure 1F). They showed focal positivity for EMA, SMA, CD15, and CD99. A few cells showed weak staining for CK8/18. Assays were negative for pan-cytokeratin, MSA, myogenin, Myo-D1, chromogranin, synaptophysin, CD34, CD31, CD117, and CA19-9. Tumor staining for WT-1 was inconclusive. The aforementioned tumor morphology along with the immunophenotype raised our suspicion for DSRCT. A molecular study for t(11;22)
Discussion
DSRCT is an uncommon sarcomatous neoplasm with a young male predilection. Most of the reported cases were found to arise in the pelvic and intra-abdominal region [5]. Although the most common site is the peritoneum, extraperitoneal DSRCT has been described in various body organs, such as the parotid gland, ovary, testis, and pleura, and orbit [2–4]. To the best of our knowledge, only 5 reported cases have described DSRCT in the pancreas as a predominant intra-abdominal solid organ involvement [6–10] (Table 1). Individuals with DSRCT present with various clinical presentations, from asymptomatic to a complex large intra-abdominal mass manifested as abdominal pain or symptoms of compression. Only one previously reported case included painful obstructive jaundice [8]. Our case is interesting as the mass was discovered incidentally after mild abdominal trauma. It subsequently progressed rapidly, and it was found to arise from the pancreas. It adhered to the spleen, splenic flexure, and part of the colon, which required aggressive surgical removal through exploratory laparotomy. In this case, the pancreas was almost replaced by the tumor. The distal part was involved initially, followed by involvement of the head of the pancreas. However, there was no evidence of pelvic, omental, or peritoneal deposits or ascites, which made a primary peritoneal DSRCT with invasion to the pancreas less likely. Histopathologic features of this tumor helped to reach the diagnosis. The malignant cells exhibited features of primitive cells with scant cytoplasm and hyperchromatic nuclei with inconspicuous nucleoli. They were arranged in variable histologic patterns and formed tubules or nests encircled by dense desmoplastic fibro-myxoid stroma. The diagnosis of this tumor is challenging in children, even with known polyphenotypic immunohistochemistry markers, because it does not have a specific radiological image and it may show various types of differentiation such as epithelial, mesenchymal, or neuronal [11]. Characteristically, DSRCT expresses desmin in a dot-like pattern, as in our case, and nuclear positivity for WT-1 along with immunoreactivity for an epithelial component favors DSRCT over other small round blue cell tumors. Unfortunately, WT-1 was inconclusive in our case. However, the current case was negative for myogenin and MyoD1, which made rhabdomyosarcoma unlikely. In addition, the negativity for chromogranin and synaptophysin helped to rule out the possibility of neuroendocrine tumors. At the time, Ewing sarcoma/PNET could not be completely excluded due to focal membranous positivity for a highly sensitive marker, CD99. However, CD99 is not specific, and many other small round blue cell tumors may express it. The diagnosis of DSRCT was confirmed by detecting a characteristic cytogenetic abnormality of gene fusion between the
Conclusions
Despite the rarity cases of DSRCT, even in the classic abdominopelvic cavity, several studies have documented DSRCT arising in a different body organs including the pancreas. Considering DSRCT within the continuum of differential diagnoses of small round blue cell tumors, particularly in extraperitoneal locations, is beneficial. However, there are no known significant differences between pancreatic compared with primary peritoneal DSRCT. Reaching a precise diagnosis of DSRCT is essential because the behavior and management of this neoplasm may differ markedly. In challenging cases, the characteristic trans-location and fusion gene (
References:
1.. Gerald WL, Rosai J, Case 2. Desmoplastic small round cell tumor with divergent differentiation: Pediatr Pathol, 1989; 9; 177-83
2.. Hatanaka KC, Takakuwa E, Hatanaka Y, Desmoplastic small round cell tumor of the parotid gland – report of a rare case and a review of the literature: Diagn Pathol, 2019; 14(1); 43
3.. Kretschmar CS, Colbach C, Bhan I, Crombleholme TM, Desmoplastic small cell tumor: Aa report of three cases and review of literature: J Paediatr Hematol Oncol, 1996; 18; 293-98
4.. He XR, Liu Z, Wei J, Primary desmoplastic small round cell tumor in the left orbit: A case report and literature review: Int Ophthalmol, 2019; 39; 471-75
5.. Gil A, Portilla AG, Brun EA, Sugarbaker PH, Clinical perspective on desmo-plastic small round-cell tumor: Oncology, 2004; 67; 231-42
6.. Bismar TA, Basturk O, Gerald WL, Desmoplastic small cell tumor in the pancreas: Am J Surg Pathol, 2004; 28; 808-12
7.. Seshadri A, Arora A, Agrawal N, Chattopaddhaya TK, Desmoplastic small round cell tumor of pancreas – a rare entity: Pancreatology, 2014; 14; S4
8.. Subasinghe D, Keppetiyagama CT, Sudasinghe H, Pancreatic desmo-plastic small round cell tumour – a rare presentation of painful obstructive jaundice: JOP, 2014; 15; 618-21
9.. Qureshi S, Shrikhande S, Ramadwar M, Desmoplastic small round cell tumor of the pancreas: An unusual primary site for an uncommon tumor: J Indian Assoc Pediatr Surg, 2011; 16; 66-68
10.. Ryan A, Razak A, Graham J, Desmoplastic small round-cell tumor of the pancreas: J Clin Oncol, 2007; 25(11); 1440-42
11.. Kim JW, Park JH, Cho HJ, A case of desmoplastic small round cell tumor diagnosed in a young female patient: Cancer Res Treat, 2009; 41; 233-36
12.. Bulbul A, Fahy BN, Xiu J, Desmoplastic small round blue cell tumor: A review of treatment and potential therapeutic genomic alterations: Sarcoma, 2017; 2017; 1278268
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report  22,759,844
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133