30 September 2020: Articles 
Late-Onset Bartter Syndrome Type II Due to a Homozygous Mutation in Gene: A Case Report and Literature Review
Unusual clinical course, Rare disease
Khaled A. Elfert1ABCDEF*, David S. Geller23ADEF, Carol Nelson-Williams4ABCDF, Richard P. Lifton4ACD, Hassan Al-Malki5AB, Awais Nauman5ABCDFDOI: 10.12659/AJCR.924527
Am J Case Rep 2020; 21:e924527






Abstract
BACKGROUND: Bartter syndrome is a rare genetic disease characterized by hypokalemia, metabolic alkalosis, and hyperreninemic hyperaldosteronism. Five different subtypes have been described based on the genetic defect identified. Bartter syndrome type II is caused by homozygous or compound heterozygous loss-of-function mutations in the KCNJ1 gene encoding ROMK. This subtype is typically described as a severe antenatal form of the disease, often presenting with polyhydramnios before childbirth.
CASE REPORT: Here, we describe the case of a 26-year-old man who presented with generalized body weakness and hypokalemia and was ultimately diagnosed with Bartter syndrome type II based on his clinical features coupled with the identification of a homozygous missense mutation in KCNJ1.
CONCLUSIONS: To the best of our knowledge, this is the fifth case of late-onset Bartter syndrome type II. Interestingly, the mutation identified in our patient has been previously described in patients with antenatal Bartter’s Syndrome. The late presentation in our patient suggests a surprising degree of phenotypic variability, even in patients carrying the identical disease-causing mutation.
Keywords: Bartter Syndrome, Hypokalemia, Mutation, Missense, Nephrocalcinosis, Potassium Channels, Inwardly Rectifying, Homozygote, Pregnancy
Background
Bartter syndrome is a rare condition with a prevalence of 1 in 1 000 000 [1]. It is characterized by polyuria and polydipsia, hypokalemia, metabolic alkalosis, and high renin and aldosterone levels.
Bartter syndrome results from gene mutations leading to impaired function of the transporters or channels responsible for sodium chloride reabsorption in the thick ascending part of the loop of Henle. Bartter syndrome type I is due to
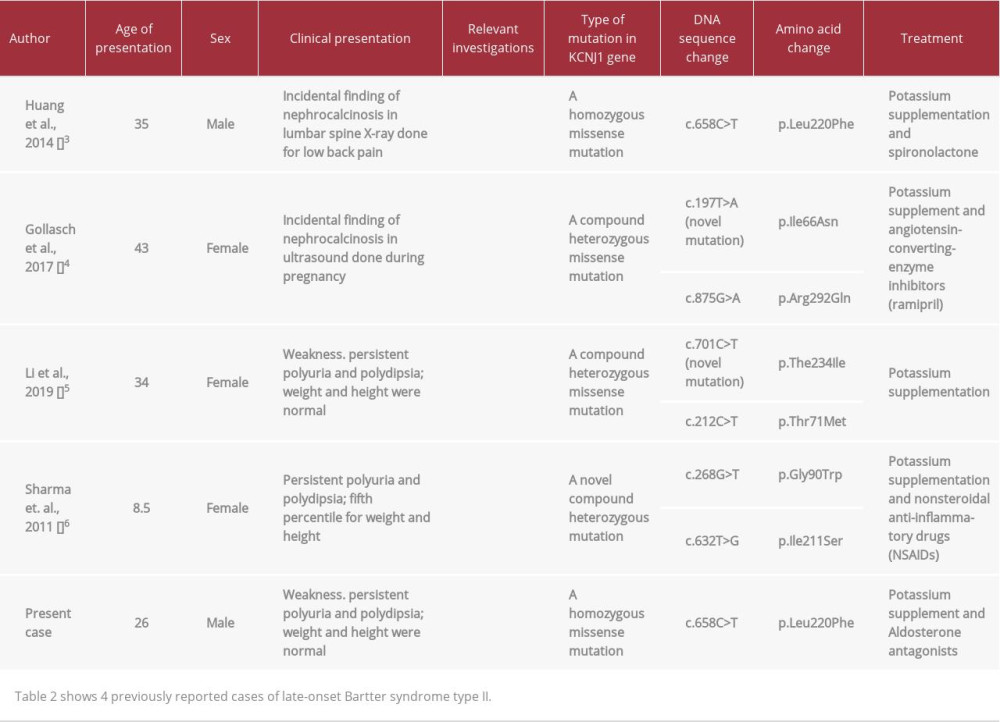
It is uncommon for type II to present in adolescence or adult life. To the best of our knowledge, there are only 4 reported cases of late-onset type II Bartter syndrome [3–6]. Here, we report the case of a patient with adult presentation of Bartter syndrome type II. The genetic mutation in our patient was identical to the mutation found in patients with neonatal presentation [7], suggesting a surprising degree of phenotypic diversity.
Case Report
GENETIC ANALYSIS:
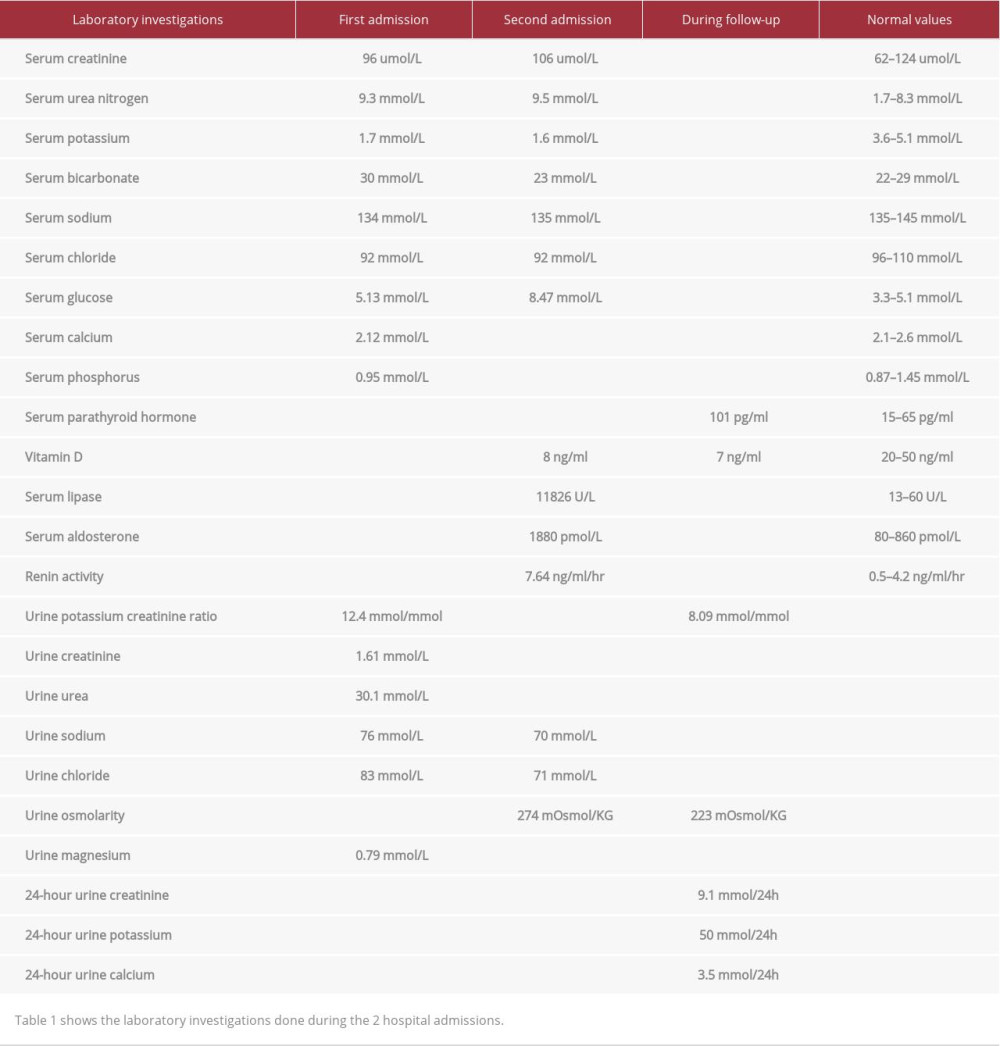
The clinical picture of recurrent hypokalemia with renal potassium wasting, elevated urine chloride, hyperreninemia, and hyperaldosteronemia, coupled with the demonstration of calcific foci in the kidney with medullary nephrocalcinosis, raised the suspicion of Bartter syndrome.
After obtaining informed consent, a blood sample was sent to the Yale University School of Medicine for genetic testing. The sample was sequenced using the forward KCNJ1 primer (Figure 1) and the reverse KCNJ1 primer. The sequencing using both primers showed a homozygous missense mutation in the KCNJ1 gene in which a cytosine nucleotide replaced a thymine nucleotide (c.658C>T), leading to a substitution of phenylalanine for leucine at codon 220 (L220F). This position is entirely conserved among species (Figure 2) and is highly conserved among paralogs (Figure 3), indicating that the position is important for functioning of this protein. Moreover, this mutation has been previously identified as a disease-causing mutation for Bartter’s syndrome. The patient’s parents and the siblings were not available for genetic analysis.
Based on the clinical picture and the genetic results, the patient was diagnosed with type II Bartter syndrome.
FOLLOW-UP:
His potassium level has remained between 3.5 and 4 mmol/L on this treatment. In 2010, he developed gynecomastia and underwent liposuction. The spironolactone was discontinued, and his potassium level decreased to 3.0 mmol/L and eplerenone 50 mg once daily was started, with normalization of his serum potassium. He maintained his potassium in the following 2 years within the normal range on eplerenone 50 mg TID and potassium chloride supplementation. His serum creatinine has ranged between 110 and 143 umol/L.
After the first episode of acute pancreatitis, the patient had multiple subsequent episodes of acute pancreatitis over years of follow-up, for which extensive workup was unrevealing. He underwent empirical cholecystectomy, but he continued to have recurrent moderate-to-severe abdominal pain requiring tramadol use. During these episodes, the pancreatic enzymes were mildly elevated, although at other times they were normal. The possibility of early chronic pancreatitis or sphincter of Oddi dysfunction was considered.
Discussion
In summary, we present the case of a man who presented with recurrent episodes of severe hypokalemia after the age of 26 years. Work-up revealed urinary potassium wasting with nephrocalcinosis, and the patient was diagnosed with Bartter syndrome type II based on the presence of a homozygous mutation in
The ROMK channel is necessary for the process of sodium chloride reabsorption in the ascending part of the loop of Henle [5].
The impaired reabsorption of sodium chloride in this part of the nephron leads to renal salt wasting and volume contraction, stimulating the renin-angiotensin-aldosterone system. In addition, the increased delivery of sodium chloride to the distal part of the nephron leads to increased secretion of potassium and hydrogen, resulting in hypokalemia and metabolic alkalosis.
The adult presentation of our patient is unusual for type II Bartter syndrome. Typically, infants diagnosed with type II have a history of polyhydramnios and premature delivery, and they present with failure to thrive, polyuria, and dehydration [6]. Unlike children with the other forms of Bartter syndrome, they have transient hyperkalemia and metabolic acidosis in the early postnatal life [7]. Finer et al. demonstrated in a case-series of 12 infants with type II Bartter syndrome that, after this period of transient hyperkalemia, the patients had normal serum potassium during their childhood, which could be the case in our patient. Similar to type I, nephrocalcinosis is a common finding in Bartter syndrome type II [6].
Our patient had hypokalemia, metabolic alkalosis, and normal blood pressure; hence, urine chloride testing was warranted to narrow the differential diagnosis. Since the urine chloride level was high, the possibilities were a renal tubular genetic defect (e.g., Bartter syndrome and Gitelman syndrome) or diuretic intake, which was denied by the patient. In addition, in multiple visits, the patient had a high potassium/creatinine ratio with concurrent hypokalemia, suggesting renal potassium wasting. Genetic testing was then performed, which revealed the diagnosis of Bartter syndrome type II.
In our literature review, we found only 4 case reports of patients with late presentation of Bartter syndrome type II; 3 of them presented in adult life. These 4 cases had several findings in common. Similar to our case, renal imaging in all the cases revealed nephrocalcinosis. In addition, all cases had hyperreninemic hyperaldosteronism, which is a common finding in all types of Bartter syndrome. Hypercalciuria is an inconsistent finding in these cases, with marked hypercalciuria in some, but normal calcium excretion in others (Table 2). The absence of hypercalciuria is curious in our patient, but likely reflects hypovitaminosis D (Table 1) and resultant decreased intestinal calcium absorption [4].
An interesting feature of our case is the phenotypic diversity that has been described for individuals homozygous for the L220F mutation. Both our patient and the patient reported by Huang et al. [3] had this mutation, and both presented as adults. In contrast, Walsh et al. described a patient homozygous for the identical mutation that presented before age 1 year [9]. Similarly, Vollmer et al. described a Turkish child, compound heterozygous for the L220F mutation, who presented as a neonate with typical Bartter’s type II symptoms [10]. Interestingly,
Our patient’s presentation is also notable for unexplained recurrent pancreatitis. ROMK is expressed in the pancreas [18,19], so we cannot exclude that the mutant ROMK may be playing a role in the disease process. We were unable to identify other case reports of pancreatitis in patients with Bartter syndrome, but we note that most such case reports highlight the cases of children. It will be of interest to determine if pancreatic phenotypes are noted if more older patients are reported.
Conclusions
Our case revealed that, in rare conditions, type II of Bartter syndrome can have an adult presentation, which is consistent with a few previous observations. L220F mutation in
Figures
References:
1.. Ji W, Foo JN, O’Roak BJ, Rare independent mutations in renal salt handling genes contribute to blood pressure variation: Nat Genet, 2008; 40(5); 592-99
2.. Cunha T da S, Heilberg IP, Bartter syndrome: Causes, diagnosis, and treatment: Int J Nephrol Renovasc Dis, 2018; 11; 291-301
3.. Huang L, Luiken GPM, van Riemsdijk IC, Nephrocalcinosis as adult presentation of bartter syndrome type II: Neth J Med, 2014; 72(2); 91-93
4.. Gollasch B, Anistan YM, Canaan-Kühl S, Gollasch M, Late-onset Bartter syndrome type II: Clin Kidney J, 2017; 10(5); 594-99
5.. Li J, Hu S, Nie Y: Medicine (Baltimore), 2019; 98(34); e16738
6.. Sharma A, Linshaw MA, A novel compound heterozygous romk mutation presenting as late onset bartter syndrome associated with nephrocalcinosis and elevated 1,25(oh) 2 vitamin d levels: Clin Exp Nephrol, 2011; 15(4); 572-76
7.. Walsh PR, Tse Y, Ashton E, Clinical and diagnostic features of Bartter and Gitelman syndromes: Clin Kidney J, 2018; 11(3); 302-9
8.. Lin SH, Lin YF, Chen DT, Laboratory tests to determine the cause of hypokalemia and paralysis: Arch Intern Med, 2004; 164(14); 1561-66
9.. Emmett M, Ellison DH, Bartter and Gitelman syndromes – UpToDate. In: UptoDate., 2018 https://uptodate.publicaciones.saludcastillayleon.es/contents/bartter-and-gitelman-syndromes?search=gitelman&source=search_result&selectedTitle=1~21&usage_type=default&display_rank=1
10.. Brochard K, Boyer O, Blanchard A, Phenotype-genotype correlation in antenatal and neonatal variants of Bartter syndrome: Nephrol Dial Transplant, 2009; 24(5); 1455-64
11.. Finer G, Shalev H, Birk OS, Transient neonatal hyperkalemia in the antenatal (ROMK defective) Bartter syndrome: J Pediatr, 2003; 142(3); 318-23
12.. Black CE, Berg RL, Urquhart AC, 24-Hour urinary calcium in primary hyper-parathyroidism: Clin Med Res, 2013; 11(4); 219-25
13.. Vollmer M, Koehrer M, Topaloglu R, Two novel mutations of the gene for Kir 1.1 (ROMK) in neonatal Bartter syndrome: Pediatr Nephrol, 1998; 12(1); 69-71
14.. Kunzelmann K, Hübner M, Vollmer M, A bartter’s syndrome mutation of ROMK1 exerts dominant negative effects on K+conductance: Cell Physiol Biochem, 2000; 10(3); 117-24
15.. Srivastava S, Li D, Edwards N: Physiol Rep, 2013; 1(6); 1-8
16.. Mijnders M, Kleizen B, Braakman I, Correcting CFTR folding defects by small-molecule correctors to cure cystic fibrosis: Curr Opin Pharmacol, 2017; 34; 83-90
17.. Shuck ME, Bock JH, Benjamin CW, Cloning and characterization of multiple forms of the human kidney ROM-K potassium channel: J Biol Chem, 1994; 269(39); 24261-70
18.. Kondo C, Isomoto S, Matsumoto S, Cloning and functional expression of a novel isoform of ROMK inwardly rectifying ATP-dependent K+ channel, ROMK6 (Kir1.1f): FEBS Lett, 1996; 399(1–2); 122-26
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133