25 November 2020: Articles 
Phenotypic Heterogeneity in 5 Family Members with the Mitochondrial Variant m.3243A>G
Challenging differential diagnosis, Rare disease, Educational Purpose (only if useful for a systematic review or synthesis)
Josef Finsterer1ABCDEF*, Franco Laccone2BCDEDOI: 10.12659/AJCR.927938
Am J Case Rep 2020; 21:e927938






Abstract
BACKGROUND: The pathogenic mitochondrial DNA variant m.3243A>G is associated with a wide range of clinical features, making disease course and prognosis extremely difficult to predict. We aimed to understand the cause of this broad intra-familial phenotypic heterogeneity in a large family carrying the variant m.3243A>G.
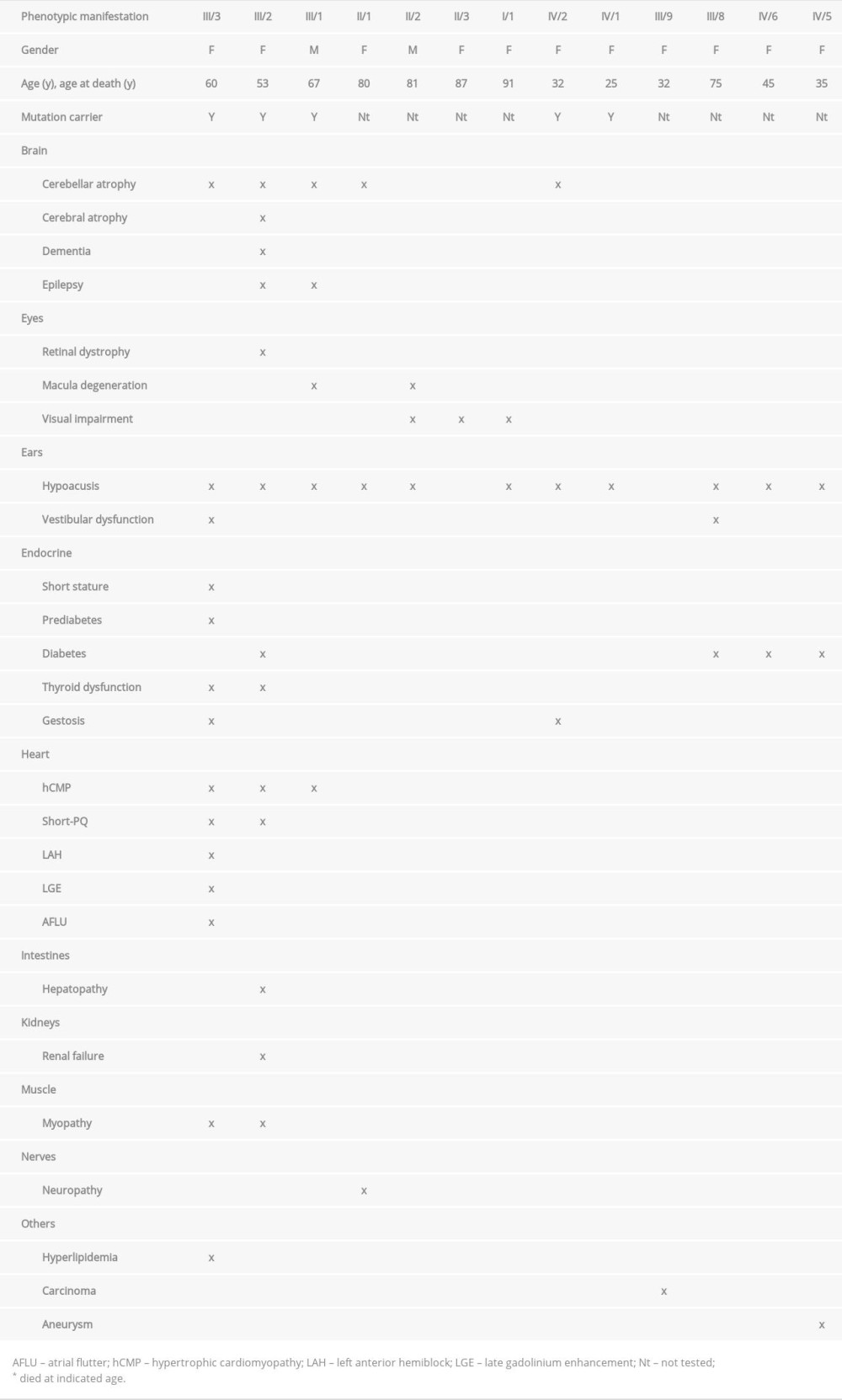
CASE REPORT: Thirteen family members were clinically affected. Clinical manifestations occurred in the brain, eyes, ears, endocrine organs, myocardium, intestines, kidneys, muscle, and nerves. Five family members carried the m.3243A>G variant. The 2 most severely affected patients were the index patient, a 60-year-old woman, and her sister, who was deceased. The phenotypic features most frequently found were hypoacusis and cerebellar atrophy. Hypertrophic cardiomyopathy was diagnosed in 3 family members. Short PQ syndrome and gestosis had not been reported to date. The broad phenotypic heterogeneity was attributed to variable heteroplasmy rates and variable mtDNA copy numbers. All affected patients benefited from symptomatic treatment.
CONCLUSIONS: The mitochondrial DNA variant m.3243A>G can manifest phenotypically with a non-syndromic, multisystem phenotype with wide intra-familial heterogeneity. Rare manifestations of the m.3243A>G variant are gestosis and short PQ syndrome. The broad intra-familial phenotypic heterogeneity may be related to fluctuating heteroplasmy rates or mitochondrial DNA copy numbers and may lead to misdiagnosis for years.
Keywords: Cardiomyopathies, MELAS syndrome, Mitochondrial Diseases, DNA, Mitochondrial, Family, heteroplasmy, Mutation, Phenotype
Background
The mitochondrial DNA (mtDNA) variant m.3243A>G manifests with syndromic or non-syndromic phenotypes [1]. Among the syndromic phenotypes, mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome is the most well-known [2]. Other syndromic phenotypes due to the variant m.3243A>G include maternally inherited diabetes and deafness (MIDD) syndrome, myoclonic epilepsy with ragged red fiber (MERRF) syndrome, Leigh syndrome, or MELAS/KSS (Kearns-Sayre syndrome) overlap syndrome [1]. Non-syndromic phenotypes commonly include multisystem disorders affecting the brain, eyes, ears, endocrine organs, myocardium, gastrointestinal tract, and the kidneys in variable combinations. Little is known about the frequency and expression of nonsyndromic m.3243A>G-related phenotypes. Here, we present a family carrying the variant m.3243A>G with a non-syndromic phenotype and broad intra-familial phenotypic heterogeneity.
Case Reports
The index patient (III/3) was a 60-year-old white woman, height 164 cm, weight 45 kg with a multisystem mitochondrial disorder (MID) initially classified as MIDD syndrome. The initial clinical manifestation was growth retardation resulting in short stature. At age 35 years, hypoacusis became apparent, requiring bilateral hearing devices. At age 39 years, ataxic gait developed, being attributed to cerebellar atrophy on imaging. At age 48 years, she underwent subtotal thyroidectomy because of struma multinodosa. Starting at age 52 years, she experienced recurrent episodes of vertigo, which later became permanent and were attributed to vestibular dysfunction.
Starting at age 55 years, slowly progressive weakness of the upper limbs and recurrent cramps of the upper and lower limb muscles developed, preventing her from sleeping properly. Additionally, she experienced easy fatigability. Starting at age 59 years, she experienced paresthesias of the distal lower limbs. Nerve conduction studies revealed axonal, sensori-motor polyneuropathy. A genetic work-up at age 57 years by means of PCR and Sanger sequencing revealed the common variant m.3243A>G in
At age 58 years she experienced acute heart failure attributed to hypertrophic cardiomyopathy (hCMP) (septum: 15 mm, posterior wall: 16 mm) with a hypokinetic septum, an ejection fraction (EF) of 25%, dilated atria, hypertension (PAP 45 mmHg), and mild mitral and tricuspid insufficiency. The proBNP value was 9508 ng/L (n, <125 ng/L). ECG showed left anterior hemiblock and a shortened PQ time (<0.12 ms) without a delta-wave (short PQ syndrome). Holter monitoring revealed recurrent, arrhythmic, supraventricular salvos. Coronary angiography was normal. Cardiac MRI, 9 months after heart failure, revealed an EF of 56%, mild septal hypertrophy (12 mm), and postero-latero-basal late gadolinium enhancement (LGE). At age 59 years, paroxysmal atrial flutter with 2: 1 transmission was diagnosed. Cardioversion was successful.
She had a history of smoking 10 cigarettes per day but stopped smoking at age 58 years. Chronic obstructive pulmonary disease (COPD)-II was additionally diagnosed. Blood chemical investigations revealed erythrocytosis, hepatopathy, and renal insufficiency (Table 1). Additionally, pre-diabetes was diagnosed, without requiring therapy.
A neurologic exam at age 60 years revealed short stature, dry eyes, hypoacusis, mild dysarthria, sore neck muscles, diffuse weakness of the upper limbs (M4), weak foot extension (M5-), diffuse wasting of the upper and lower limbs, absent tendon reflexes on the upper and lower limbs, and gait ataxia.
She was regularly taking bisoprolol (5m g/day), lisinopril (5 mg/day), furosemide (20 mg/day), edoxaban (30 mg/day), and tiotropium bromide. She was drinking approximately 500 mL/d. She was recommended to additionally take coenzyme-Q (300 mg/day).
The family history was positive for multisystem MID in her mother (II/1), sister (III/2), and brother (III/1); in the sister (II/3) and brother (II/2) of the mother (Table 1); and in her 2 daughters (IV/1, IV/2), 2 female cousins (III/8, III/9), and 2 nieces (IV/5, IV/6) (Figure 1). In 5 of these clinically affected relatives, the m.3243A>G variant was detected (Figure 1). The phenotype varied considerably among these family members (Table 1). The mother of the index patient had died at age 80 years from pulmonary embolism. During adulthood she had developed hypoacusis, cerebellar ataxia, and polyneuropathy. One brother of the mother, aged 81 years, experienced hypoacusis since age 50 years and macula dystrophy. He had 3 healthy children (2 girls, 1 boy). The sister of the mother (II/3) had a suspected visual problem and died at age 87 years. The grandmother from the mother’s side (I/1) had a history of visual impairment and hearing loss and died at age 91 years. The sister of the index patient (III/2) (height 155 cm, weight 42 kg), had epilepsy, diffuse cerebral atrophy with dementia, cerebellar atrophy with ataxia, leucoencephalopathy, hypoacusis requiring cochlear implants, retinal dystrophy, diabetes (HbA1c up to 12.1), hyperlipidemia, struma nodosa requiring thyroidectomy, hCMP with intermittent short PQ syndrome and mild aortic and mitral insufficiency, hepatopathy, muscle weakness with hyper-CKemia, a single pancreatic cyst, surgery for squinting as a child, rectal discharge, near-drowning because of muscle weakness at age 62 years, and renal insufficiency.
She carried the m.3243A>G variant with a heteroplasmy rate of 40–50% in buccal mucosa cells. She died at age 53 years from heart failure and renal insufficiency. The brother (III/1), aged 67 years, had a history of epilepsy, hypoacusis, macular dystrophy (fundus flavimaculatus), and hCMP (Table 1). The 2 daughters of the index patient (IV/1, IV/2) also carried the m.3243A>G variant. The older daughter (IV/2), now aged 32 years, manifested with cerebellar atrophy, hearing loss, and gestosis requiring cesarian section in gestational week 29. Her son (V/1) was born by cesarean section in gestational week 25 and presented with mild delayed motor development. The younger daughter of the index patient (IV/1), aged 25 years, complained of impaired hearing and tinnitus. The index patient has 4 female cousins. The oldest cousin (III/8), aged 75 years, has diabetes, hearing impairment, and vestibular dys-function. The second cousin (III/9) died from cancer at age 32 years after the birth of her fourth child (2 boys, 2 girls). One girl, now age 45 years (IV/6), has diabetes and hypoacusis. The second girl (IV/5) had diabetes and hypoacusis and died from aneurysmal bleeding at age 35 years. The 2 other cousins are healthy.
Discussion
The presented family is interesting for multisystem MID due to the variant m.3243A>G in 5 family members, manifesting with a non-syndromic phenotype with high intra-familial variability. MELAS was excluded as none of the family members carrying the variant manifested with a stroke-like episode (SLE). None of the mutation carriers fulfilled the Japanese or Hirano-criteria [3,4] for diagnosing MELAS. MIDD was excluded, as only 1 of the mutation carriers had deafness and diabetes together. The index patient was diagnosed only with pre-diabetes, which never required anti-diabetic medication. MERRF was excluded, as none of the mutation carriers fulfilled the diagnostic criteria for MERRF [5]. Leigh syndrome was excluded based on the non-compatible clinical presentation and absence of typical features on cerebral MRI [6]. None of the mutation carriers fulfilled the diagnostic criteria for KSS. Based on these considerations, a non-syndromic MID with wide phenotypic variability was diagnosed [7].
The index patient (III/3) manifested in the brain, eyes, ears, endocrine organs, heart, intestines, kidneys, muscle, and nerves (Table 1). Her sister (III/2) manifested in the brain, ears, endocrine organs, liver, muscle, kidneys, and heart (Table 1). Interestingly, the index patient developed gestosis during both of her pregnancies. The brother of the index patient (III/1) manifested clinically in the brain, eyes, ears, and heart (Table 1). The older daughter of the index patient (IV/2) manifested in the brain and ears (Table 1). Her sister (IV/2) manifested only in the ears with tinnitus and mild hypoacusis. The grandson of the index patient (V/1) became apparent because of motor developmental delay. One uncle from the mother’s side (II/2) manifested in the ears, brain, and eyes (Table 1). An aunt from the mother’s side (II/3) possibly manifested in the eyes. Two of the index patient’s nieces (IV/5, IV/6) manifested in the ears and the endocrine organs. A female cousin of the index patients (III/9) manifested with a carcinoma. Of her 4 children, 2 were affected (IV/5, IV/6) and manifested in the ears and the endocrine organs.
The phenotypic features that were most frequently observed in this family include deafness and cerebellar atrophy (Table 1). Previously unreported phenotypic features in m.3243A>G carriers were gestosis and short PQ syndrome (Table 1). Rarely described phenotypic features included LGE and psychomotor retardation. LGE has been reported only once [8]. However, in a study of 22 m.3243A>G mutation carriers by cardiac MRI, none of the included patients showed LGE [9]. Three probands had hCMP. All other features occurred only in 1 or 2 probands. Hyperlipidemia was considered an m.3243A>G manifestation, as MID patients frequently manifest with hereditary disturbance of lipid metabolism. The phenotypic heterogeneity could be also due to the fact that disease duration varied significantly among family members. However, there were some probands with similar disease duration but variable pheno-type. The phenotypic heterogeneity may not only be due to heterogeneous heteroplasmy rate, but could be also due to variable copy numbers within a mitochondrion. Unfortunately, copy numbers were not counted in any of the family members. Recently, nuclear factors were identified explaining some of the phenotypic divergence [10]. The low heteroplasmy rates in blood and buccal mucosa cells do not exclude pathogenicity, as they were determined in tissues that are largely unaffected clinically.
Conclusions
This case study shows that the mtDNA variant m.3243A>G can manifest phenotypically in a non-syndromic way with large intra-familial phenotypic heterogeneity. So far, unreported manifestations of the m.3243A>G variant were gestosis and intermittent short PQ syndrome. The broad intra-familial variability may be related to the variable heteroplasmy rates in affected or non-affected tissues, to the phenotypic expression, which modifies the disease course, and the diet, drugs, environmental poisoning, and lifestyle of mutation carriers. Acronyms for mitochondrial syndromes may help to classify a patient but do not contribute much to the understanding of the phenotypic variability of mtDNA variants. The broad phenotypic variability may lead to misdiagnosis for years.
References:
1.. Finsterer J, Zarrouk-Mahjoub S, The heart in m.3243A>G carriers: Herz, 2020; 45; 356-61
2.. El-Hattab AW, Almannai M, Scaglia F: MELAS Feb 27, 2001; 1993-2020, GeneReviews® Seattle (WA), University of Washington Seattle http://www.ncbi.nlm.nih.gov/books/NBK1233/
3.. Hirano M, Ricci E, Koenigsberger MR, Melas: An original case and clinical criteria for diagnosis: Neuromuscul Disord, 1992; 2; 125-35
4.. Yatsuga S, Povalko N, Nishioka J, MELAS: A nationwide prospective cohort study of 96 patients in Japan: Biochim Biophys Acta, 2012; 1820; 619-24
5.. Finsterer J, Zarrouk-Mahjoub S, Shoffner JM, MERRF Classification: Implications for diagnosis and clinical trials: Pediatr Neurol, 2018; 80; 8-23
6.. Finsterer J, Leigh and Leigh-like syndrome in children and adults: Pediatr Neurol, 2008; 39; 223-35
7.. Liu G, Shen X, Sun Y, Heteroplasmy and phenotype spectrum of the mitochondrial tRNALeu (UUR) gene m.3243A>G mutation in seven Han Chinese families: J Neurol Sci, 2020; 408; 116562
8.. Jose T, Gdynia HJ, Mahrholdt H, CMR gives clue to “ragged red fibers” in the heart in a patient with mitochondrial myopathy: Int J Cardiol, 2011; 149; e24-27
9.. Bates MG, Hollingsworth KG, Newman JH, Concentric hypertrophic remodelling and subendocardial dysfunction in mitochondrial DNA point mutation carriers: Eur Heart J Cardiovasc Imaging, 2013; 14; 650-58
10.. Pickett SJ, Grady JP, Ng YS, Phenotypic heterogeneity in m.3243A>G mitochondrial disease: The role of nuclear factors: Ann Clin Transl Neurol, 2018; 5; 333-45
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952428
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952854
Most Viewed Current Articles
07 Dec 2021 : Case report  22,760,204
22,760,204
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,117
176,117
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,604
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,593
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133