23 November 2020: Articles 
Cerebellar Vertigo as an Unspecific Initial Presentation of Creutzfeldt-Jakob Disease
Rare disease
Maham Mehmood1ABCDEF, Haider Ghazanfar1ABCDEF*, Manjeet S. Dhallu2ABCDEF, Eghosa Omoregie2ABCDEFDOI: 10.12659/AJCR.927955
Am J Case Rep 2020; 21:e927955






Abstract
BACKGROUND: Creutzfeldt-Jakob disease (CJD) is a human prion disease characterized by severe and rapidly progressive fatal neurodegeneration. Currently, there is no cure for CJD, and death from CJD usually occurs within 1 year from the onset of the symptom, and the median survival time is 6 months.
CASE REPORT: The patient was a 63-year-old woman who presented to the hospital reporting having vertigo for the past 1 week and involuntary muscle contraction resulting in slow repetitive movement and abnormal posture for the past 3 days. A physical examination at the time of admission revealed unsteady gait, dystonia, and dysmetria of the left upper limb. There was no nystagmus on examination. Electroencephalography done on the same day showed focal epileptiform discharge on bilateral temporal lobes, which were more on the right side than the left. It also showed mild diffuse cerebral slowing. Cerebrospinal fluid analysis showed positive for RT-QulC, T tau protein, and 14-3-3. A diagnosis of CJD was made based on clinical course, imaging, and cerebrospinal fluid analysis.
CONCLUSIONS: The diagnosis of CJD can be suspected based on clinical signs and symptoms and can be confirmed after performing MRI, EEG, and lumbar puncture. Therefore, it is important to recognize vertigo as an unspecific symptom of CJD so that a timely diagnosis can be made and unnecessary procedures can be avoided.
Keywords: Creutzfeldt-Jakob Syndrome, Diagnosis, Morbidity, Mortality, Neurodegenerative Diseases, Electroencephalography, Magnetic Resonance Imaging, Spinal Puncture, Vertigo
Background
Creutzfeldt-Jakob disease (CJD) is a human prion disease characterized by severe and rapidly progressive fatal neurodegeneration. According to a multinational study, the worldwide annual mortality rate of sporadic CJD was 1.39 per million [1].
It is characterized into 3 categories: sporadic, genetic, and acquired [2]. The acquired cases of CJD are further differentiated into iatrogenic CJD and variant CJD. Sporadic CJD accounts for 90% of all cases of sporadic prion disease [3]. The mean age of onset for the disease is between 60 and 69 years [4,5]. There is no significant difference in prevalence between males and females.
Sign and symptoms of CJD include extrapyramidal signs, signs of corticospinal tract involvement, cerebellar manifestation, myoclonus, and neuropsychiatric symptoms. Mental impairment progressively worsens with time. Currently, there is no effective cure for the disease and death usually occurs within 1 year after onset of symptoms [6].
CJD can also present with unspecific symptoms that can lead to a misdiagnosis or delay in diagnosis. We present the case of a 63-year-old woman who presented to the hospital due to vertigo and involuntary muscle contraction resulting in slow repetitive movement and abnormal posture.
Case Report
The patient was a 63-year-old woman who presented to the hospital reporting she had been experiencing vertigo for the past week and had involuntary muscle contraction resulting in slow repetitive movement and abnormal posture for the past 3 days. The patient stated that her symptoms had progressively worsened with time. She had been seen in the clinic for vertigo 1 week before. Results of a physical examination at the clinic visit were unremarkable and a computerized tomography (CT) scan of the head was negative. The patient was advised to take meclizine. She stated that she had been depressed for the past year since the death of her husband and was not seeing any doctor for depression. She denied any other significant past medical or surgical history. The patient presented to the hospital because of worsening of symptoms. She denied smoking, alcohol abuse, and use of illicit drugs. She had no apparent risk factors for acquired prion disease, and there was no family history of prion disease.
The patient was found to have stable vital signs at presentation. A physical examination at the time of admission revealed unsteady gait, dystonia, and dysmetria of the left upper limb. There was no nystagmus on examination. The patient was alert to time, place, and person. Results of a complete blood count (CBC), comprehensive metabolic profile (CMP), lipid profile, thyroid profile, coagulation panel, C-reactive protein (CRP), and erythrocyte sedimentation rate (ESR) were normal. A chest X-ray was negative for any acute pathology. A repeat CT head was negative.
The patient underwent CT angiography of the head and neck, which ruled out major vessel stenosis and occlusion but revealed a possible right posterior communicating artery. She was admitted to the hospital for further workup and management.
Psychiatry was consulted because of her depression. The patient was started on cognitive behavioral therapy and was advised to follow up with the psychiatry outpatient service after discharge from the hospital. Conversion disorder and depression were ruled out as a cause of her symptoms. Neurology was consulted. The neurology team advised us to perform magnetic resonance imaging (MRI) of the head with and without contrast.
The patient developed altered mental status on day 3 of admission. She was started on intravenous dexamethasone, ceftriaxone, vancomycin, ampicillin, and acyclovir for a suspected diagnosis of meningitis and viral encephalitis. She was also started on levetiracetam because of subclinical seizures. Lyme disease, Wilson disease, and syphilis were ruled out. Electroencephalography (EEG) done on the same day showed focal epileptiform discharge in bilateral temporal lobes, more on the right side than the left. It also showed mild diffuse cerebral slowing. EEG was repeated on day 7 of admission, which showed focal periodic sharp waves over the parietal region and severe slowing of cerebral function. An MRI of the head showed cortical ribboning and subtle diffusion-weighted imaging abnormality of gray-matter structures at the bilateral occipital, parietal, and right frontal lobe, with fluid-attenuated inversion recovery (FLAIR) hyperintensity in the right frontal cortex. There was no decreased signal on apparent diffusion coefficient (ADC). See Figure 1.
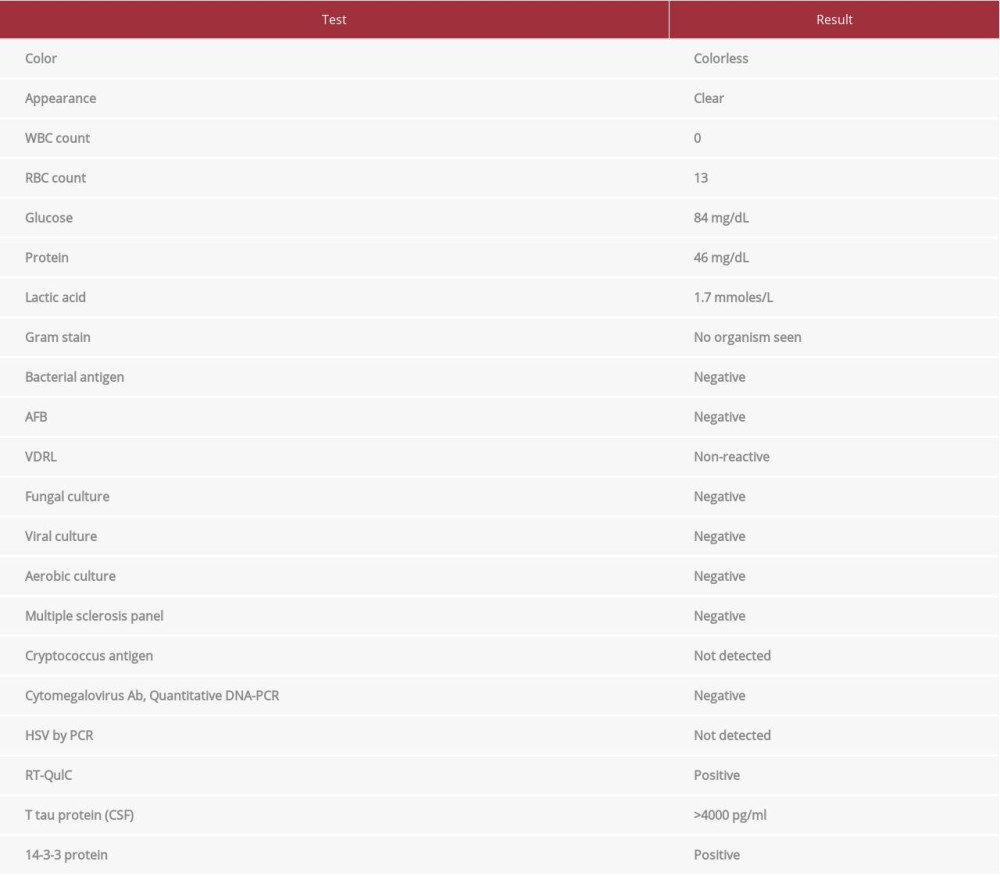
The patient underwent lumbar puncture, and cerebrospinal fluid (CSF) was sent for analysis, which ruled out meningitis and encephalitis. CSF was negative for N-Methyl- d-aspartic acid, or N-Methyl- d-aspartate (NMDA) antibody, as well as ampa1, ampa2, casper2, gaba2, hu, yo, ri, ma2, and LGI1 antibodies. CSF analysis was positive for RT-QulC, T tau protein, and 14-3-3 (Table 1).
Genetic testing was not done. The diagnosis of CJD was based on clinical, imaging, and laboratory findings. The patient had a rapid neurological decline and developed akinetic mutism. Her hospital course was complicated with acute hypoxic respiratory failure, and she was intubated. Multiple attempts to extubate her were unsuccessful. The patient underwent tracheostomy and percutaneous endoscopic gastrostomy. She was later discharged to the inpatient hospice unit, where she died. Autopsy was not done as per family wishes.
Discussion
Patients with CJD can present with a myriad of symptoms. Our patient initially presented with vertigo and later developed dystonia, dysmetria, unsteady gait, altered mental status, and akinetic mutism. According to a meta-analysis, only 2.6% of CJD patients initially presented with dizziness and vertigo [7]. Patients younger than 50 years of age were more likely to present with dizziness and vertigo as compared to patients older than 50 years of age (
The suspected diagnosis of CJD is based on clinical signs and symptoms. Once CJD is suspected, various tests can be done to confirm the diagnosis of CJD. MRI is one of the most useful modalities in diagnosing CJD because it can rule out possible causes of progressive neurological decline. A multi-center collaborative study concluded that the characteristic MRI findings seen in CJD is high signal intensity in fluid-attenuated inversion recovery (FLAIR) or diffusion-weighted imaging (DWI) in at least 2 cortical regions or signal increase in the caudate nucleus and putamen [8]. The same study found that MRI findings are considered to have the same level of diagnostic importance as that of 14-3-3 protein detection in CSF and periodic sharp wave complexes on the EEG. CT scans of the head are generally normal in CJD and help rule out other possible diagnoses.
The characteristic EEG pattern of periodic synchronous bior triphasic periodic sharp wave complex is seen in about 64% of patients with CJD [9]. CSF analysis usually reveals no cells and normal glucose levels. Some patients have elevated CSF protein levels. Real-time quaking-induced conversion (RT-QuIC) is a highly sensitive and specific test for diagnosing CJD [10]. Detection of 14-3-3 and tau protein in the CSF help diagnose CJD.
Postmortem neuropathological examination of the brain tissue is the definitive diagnostic test for CJD. As there is no effective cure of the disease, there is no therapeutic benefit in performing such an invasive procedure to confirm the diagnosis of CJD. Brain biopsy is usually considered in patients in whom a potentially treatable alternative diagnosis is suspected [11]. Death from CJD usually occurs within 1 year from the onset of the symptom, and the median survival time is 6 months [12]. Effective counseling of the patient and involvement of social services are key to managing these patients. Symptomatic management is provided to alleviate the debilitating symptoms of CJD.
Conclusions
The diagnosis of CJD can be suspected based on clinical signs and symptoms and can be confirmed by MRI, EEG, and lumbar puncture. Therefore, it is important to recognize vertigo as an unspecific symptom of CJD so that a timely diagnosis can be made and unnecessary procedures can be avoided. Early diagnosis will also help prevent iatrogenic transmission of CJD.
References:
1.. Ladogana A, Puopolo M, Croes EA, Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada: Neurology, 2005; 64(9); 1586-91
2.. Chen C, Dong XP, Epidemiological characteristics of human prion diseases: Infect Dis Poverty, 2016; 5(1); 47
3.. Puoti G, Bizzi A, Forloni G, Sporadic human prion diseases: Molecular insights and diagnosis [published correction appears in Lancet Neurol, 2012; 11(10): 841]: Lancet Neurol, 2012; 11(7); 618-28
4.. Gao C, Shi Q, Tian C, The epidemiological, clinical, and laboratory features of sporadic Creutzfeldt-Jakob disease patients in China: Surveillance data from 2006 to 2010: PLoS One, 2011; 6(8); e24231
5.. Imran M, Mahmood S, An overview of human prion diseases: Virol J, 2011; 8; 559
6.. Mackenzie G, Will R, Creutzfeldt-Jakob disease: Recent developments: F1000Res, 2017; 6; 2053
7.. Appleby BS, Appleby KK, Rabins PV, Does the presentation of Creutzfeldt-Jakob disease vary by age or presumed etiology? A meta-analysis of the past 10 years: J Neuropsychiatry Clin Neurosci, 2007; 19(4); 428-35
8.. Zerr I, Kallenberg K, Summers DM, Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease [published correction appears in Brain, 2012; 135(Pt 4): 1335]: Brain, 2009; 132(Pt 10); 2659-68
9.. Steinhoff BJ, Zerr I, Glatting M, Diagnostic value of periodic complexes in Creutzfeldt-Jakob disease: Ann Neurol, 2004; 56(5); 702-8
10.. McGuire LI, Peden AH, Orrú CD, Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease: Ann Neurol, 2012; 72(2); 278-85
11.. Zerr I, Hermann P, Diagnostic challenges in rapidly progressive dementia: Expert Rev Neurother, 2018; 18(10); 761-72
12.. Pocchiari M, Puopolo M, Croes EA, Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies: Brain, 2004; 127(Pt 10); 2348-59
In Press
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133