02 February 2021: Articles 
Four Cases of Maturity Onset Diabetes of the Young (MODY) Type 5 Associated with Mutations in the Hepatocyte Nuclear Factor 1 Beta (HNF1B) Gene Presenting in a 13-Year-Old Boy and in Adult Men Aged 33, 34, and 35 Years in Poland
Challenging differential diagnosis, Unusual or unexpected effect of treatment, Diagnostic / therapeutic accidents, Congenital defects / diseases, Educational Purpose (only if useful for a systematic review or synthesis)
Rafał Motyka1ABCDEF, Marcin Kołbuc2ABCDEF*, Wojciech Wierzchołowski3BCD, Bodo B. Beck4CDE, Iwona Ewa Towpik5BCD, Marcin Zaniew6ABCDEFGDOI: 10.12659/AJCR.928994
Am J Case Rep 2021; 22:e928994






Abstract
BACKGROUND: Maturity onset diabetes of the young (MODY) usually presents in patients under the age of 25 years and is an autosomal dominant condition associated with mutations in the hepatocyte nuclear factor 1 alpha gene, glucokinase gene, or hepatocyte nuclear factor 4 alpha gene. This report is of a series of 4 cases from Poland of MODY type 5 associated with mutations in the hepatocyte nuclear factor 1 beta (HNF1B) gene, including a 13-year-old boy and adult men aged 33, 34, and 35 years.
CASE REPORT: Three cases were diagnosed late, in patients in their mid-thirties. In two patients, the initial presentation was symptomatic diabetes complicated by ketoacidosis and hyperglycemic hyperosmolar state. Renal cysts were found in all patients, and pancreatic hypoplasia in 3 patients. All patients except 1 were negative for autoantibodies; 1 presented with hypomagnesemia. Insulin therapy was instituted in all cases. The combination of family history, imaging study results, and biochemical characteristics led to the decision to perform genetic analysis, which was conducted in 2 cases at diagnosis, and in the 2 remaining patients at 1 month and 2 years after diagnosis, respectively. Follow-up data revealed hypomagnesemia and/or hypermagnesuria in all patients.
CONCLUSIONS: We present 3 young men over 25 years and 1 boy with HNF1B-MODY. Although rare, autosomal dominant gene associations should be considered in young patients with diabetes who present with renal/pancreatic anomalies and low serum magnesium. Unusual presentation and the presence of autoantibodies should not eliminate the possibility of a HNF1B defect.
Keywords: case reports, Diabetes Mellitus, Hepatocyte Nuclear Factor 1-beta, Magnesium, Adolescent, Diabetes Mellitus, Type 2, Hepatocyte Nuclear Factor 1-alpha, Mutation, Poland
Background
Maturity onset diabetes of the young (MODY) is a rare disease, but constitutes the most common type of monogenic diabetes, with an estimated prevalence of 1 in 10 000 in adults [1]. Systematic population screening of the UK pediatric diabetes population identified 2.5% are MODY cases [2]. Despite ever-growing knowledge and clinician awareness of MODY, late diagnosis and misdiagnosis as type 2 or type 1 diabetes are not uncommon [3]. An autosomal dominant condition associated with a secretory defect of pancreas beta cells, MODY usually presents in patients below the age of 25 years [4]. As summarized by Peixoto-Barbosa et al, there are 14 known genes in which mutations result in MODY, of which the hepatocyte nuclear factor 1 alpha (HNF1A), glucokinase (
Because
This report is of a series of 4 cases of MODY5 associated with mutations in the
Case Reports
CASE 1:
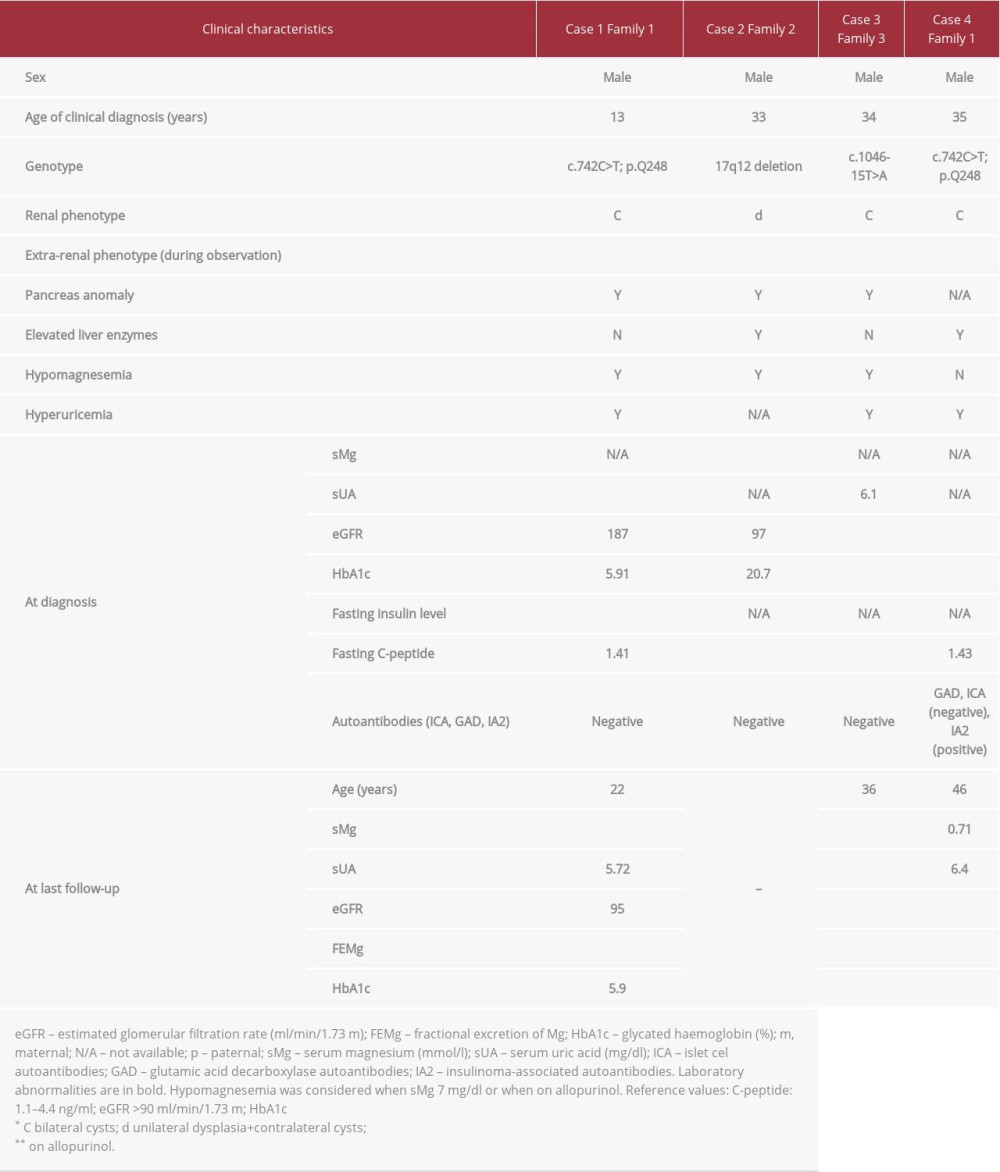
A 13-year-old boy was referred to the pediatric department for evaluation owing to fasting hyperglycemia. A glucose level of 216 mg/dL (after 2 h) during an oral glucose tolerance test confirmed the diagnosis of diabetes. The patient was born with asymptomatic spina bifida occulta of the lumbo-sacral region, posterior urethral valves, and a left pelviureteric junction obstruction. At the age of 21 months, he underwent surgical treatment for these conditions. During a thorough work-up, diabetes mellitus, hypertension, severe hypomagnesemia, hyperuricemia, proteinuria, and hypercholesterolemia were discovered. There were no islet cell antibodies (ICA), glutamic acid decarboxylase (GAD) antibodies, or insulinoma-associated (IA-2) autoantibodies found. The patient’s renal function was preserved. Ultrasound examination showed bilateral cortical renal cysts. The patient’s family history was positive for diabetes on the father’s side: the father (case 4) and grandmother, who both also had renal cysts, and 3 other relatives. The combination of renal cysts, diabetes, and positive family history raised a suspicion of
During consecutive follow-up, an abdominal computed tomography (CT) was carried out and pancreatic body and tail agenesis was noted (Figure 1). In addition, hypomagnesemia with hypermagnesuria and features of metabolic syndrome were discovered. The patient’s renal function was stable. He was initially treated with metformin, but, owing to poor glycemic control of diabetes, insulin was added. Because of the patient’s hyperuricemia and hypertension, he is being treated with allopurinol, bisoprolol, and ramipryl.
CASE 2:
A 33-year-old man was admitted to the emergency department and was found to have diabetic ketoacidosis (serum glucose of 600 mg/dL). He had been experiencing symptoms of diabetes mellitus (polyuria, polydipsia, polyphagia, and weight loss of 20 kg) over the previous 3 months. He had been addicted to alcohol for 10 years. Abdominal ultrasound and CT scans showed an atrophic left kidney and numerous cysts in the right kidney. Pancreatic calcification and agenesis of the pancreas tail were also found on the same examination. The patient’s family history was positive for diabetes in the father, who had an unspecified urinary tract malformation, and paternal grandfather. A single panel of laboratory tests taken with the patient in stable condition showed slightly elevated aminotransferase activity (ALT 69 U/L, AST 69 U/L), marked hypomagnesemia, and normal renal function. There were no ICA, GAD, or IA-2 autoantibodies found. The association of renal and pancreatic anomalies in the setting of diabetes prompted us to perform
CASE 3:
A 34-year-old man was referred for diagnostic evaluation because of significant weight loss over a 12-month period. During this work-up, hyperglycemia (serum glucose of 300 mg/dL) was discovered and a diabetes mellitus diagnosis was made. An ultrasound examination showed bilateral renal cysts with moderate impairment of renal function, corresponding to chronic kidney disease (CKD) stage II (eGFR 62 mL/min/1.73 m2). A positive family history of diabetes in the patient’s grandparents was noted. Three months later, the patient presented with jaundice and significantly elevated liver function tests (LFTs) results (total bilirubin 10.8 mg/dL, ALT 3333 U/L, AST 1559 U/L, GGTP 384 U/L, and ALP 135 U/L). Of note, a high level of testosterone and suppressed LH and FSH were disclosed.
Results of a viral hepatitis screen and antinuclear antibody test were negative. There were no evident changes in the liver on the abdominal ultrasound and CT (the liver was slightly enlarged). Oral prednisone was initiated, following which the LFTs started to normalize, and the jaundice subsided within a few weeks. This event was hypothesized to be due to anabolic steroid abuse.
As a part of the differential diagnostics, a contrast-enhanced abdominal CT was carried out. This revealed agenesis of the pancreas body and tail and confirmed renal cysts localized in both kidneys (Figure 1). On hospitalization, serum Mg2+ (sMg) was assessed for the first time, and hypomagnesemia was observed. There were no ICA, GAD, or IA-2 autoantibodies found. One month after his first presentation, the patient’s daughter was diagnosed with HNF1B nephropathy, which reverse guided the molecular diagnosis of HNF1B-MODY in our patient. A novel heterozygous mutation (c.1046-15T>A) was found in the HNF1B gene. During the follow-up period, the patient was recurrently seen on the ward because of severe abdominal pain. Porphyria was excluded. The patient was diagnosed with gastroesophageal reflux. The symptoms were relieved following treatment with a proton pomp inhibitor. On the last follow-up analysis, laboratory tests showed CKD progression to stage III, hyperuricemia, hypomagnesemia, and profound hypermagnesuria. The patient’s renal ultrasound image was stable. Intensive functional insulin therapy was initiated.
CASE 4:
A 35-year-old man presented with peripheral neuropathy. After clinical evaluation, he was diagnosed as being in a hyperglycemic hyperosmolar state. His blood glucose was >1000 mg/dL.
No diabetic ketoacidosis was present. Autoantibody screening revealed IA-2 autoantibodies, while results for ICA and GAD autoantibodies were negative. In addition, the patient was found to have hyperuricemia and hypertension. Ultrasound imaging showed bilateral hyperechogenic kidneys with single renal cysts. The patient’s renal function was moderately compromised (eGFR of 69 mL/min/1.73 m2). The underlying cause of diabetes remained unclear for 2 years until the patient’s son (case 1) was diagnosed with a
Discussion
The diagnosis of MODY5 is a challenge for clinicians. A widespread availability of molecular diagnostics permits differential diagnoses, but cost and turnaround time of larger gene panels or diagnostic exomes is an issue in many national health systems. Targeted diagnostic approaches are comparatively inexpensive and fast, with no risk of additional genetic findings that require medical actions and constitute ethical problems. In this respect, an appropriate selection of those patients that likely have a high diagnostic yield for a targeted genetic test is crucial. Timely and accurate diagnosis offers opportunities for genetic counseling, early intervention, and implementation of a coordinated care plan. Here, we presented 4 cases of
A detailed literature review by Mateus et al [10] summarizes the latest information on the role of
Early reports have described an association between
Considering the heterogeneous phenotype of
Diagnosis of MODY5 is an even greater challenge for clinicians with regards to its prevalence, inheritance, and clinical picture. It is widely accepted that a positive family history for diabetes and renal anomaly or CKD could be critical for considering
Regarding the biochemical markers, in contrast to type 1 diabetes, MODY is characterized by the absence of islet auto-antibodies, including GAD and IA-2 antibodies and detectable serum C-peptide. McDonald et al [20] observed that the prevalence of GAD and IA-2 in patients with MODY is the same as in control participants (<1%). Of note, finding islet autoantibodies, especially IA-2 antibodies, makes the diagnosis of MODY unlikely, with a negative likelihood ratio for identifying MODY from type 1 diabetes of 0.01 [21]. Thus, authors recommend molecular testing only if the clinical phenotype strongly suggests MODY rather than type 1 diabetes. Interestingly, our patient 4, who presented with IA-2 autoantibodies, is a rare MODY antibody-positive example in which renal anomalies and positive family history of diabetes supported genetic testing. With regard to serum C-peptide, its detectable level is observed in MODY and type 1 diabetes patients within the initial honeymoon period, which limits its use as a MODY predictor at first presentation. The aforementioned findings agree with the Mateus et al [10] case report, in which MODY5 was confirmed in 15-year-old girl who presented with symptomatic hyperglicemia (no ketosis). Results of a detailed clinical evaluation were negative for GAD, IA-2, and anti-zinc transporter 8 autoantibodies and a C-peptide level was detectable. Because of early antibody-negative diabetes development, concurrent renal hypoplasia, bilateral diminished corticomedullary differentiation, simple renal cysts, and positive family history of congenital anomalies of the kidney and urinary tract,
One of the underestimated biochemical hallmarks suggestive of
Conclusions
This report presents 3 young men over 25 years and one 13-year-old boy with MODY5 associated with mutations in the
References:
1.. , MODY, 2020 . Orpha.net
2.. Shepherd M, Shields B, Hammersley S, Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the U.K. pediatric diabetes population with monogenic diabetes: Diabetes Care, 2016; 39(11); 1879-88
3.. Shields BM, Hicks S, Shepherd MH, How many cases are we missing?: Diabetologia, 2010; 53(12); 2504-8
4.. Gat-Yablonski G, Shalitin S, Phillip M, Maturity onset diabetes of the young – review [published correction appears in Pediatr Endocrinol Rev,2007;5(1): 470]: Pediatr Endocrinol Rev, 2006; 3(Suppl. 3); 514-20
5.. Peixoto-Barbosa R, Reis A, Giuffrida F, Update on clinical screening of maturity-onset diabetes of the young (MODY): Diabetol Metab Syndr, 2020; 12; 50
6.. Hattersley AT, Greeley SAW, Polak M, ISPAD Clinical Practice Consensus Guidelines 2018: The diagnosis and management of monogenic diabetes in children and adolescents: Pediatr Diabetes, 2018; 19(Suppl. 27); 47-63
7.. Tattersall RB, Fajans SS, A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people: Diabetes, 1975; 24(1); 44-53
8.. Bellanne-Chantelot C, Clauin S, Chauveau D, Large genomic rearrangements in the hepatocyte nuclear factor-1 (tcf2) gene are the most frequent cause of maturity-onset diabetes of the young type 5: Diabetes, 2005; 54(11); 3126-32
9.. Bockenhauer D, Jaureguiberry G, HNF1B-associated clinical phenotypes: The kidney and beyond: Pediatr Nephrol, 2015; 31(5); 707-14
10.. Mateus JC, Rivera C, O’Meara M, Maturity-onset diabetes of the young type 5 a MULTISYSTEMIC disease: a CASE report of a novel mutation in the HNF1B gene and literature review: Clin Diabetes Endocrinol, 2020; 6; 16
11.. Sztromwasser P, Michalak A, Małachowska B, A cross-sectional study of patients referred for HNF1B-MODY genetic testing due to cystic kidneys and diabetes: Pediatr Diabetes, 2020; 21(3); 422-30
12.. Faguer S, Chassaing N, Bandin F, The HNF1B score is a simple tool to select patients for HNF1B gene analysis: Kidney Int, 2014; 86(5); 1007-15
13.. Kołbuc M, Leßmeier L, Salamon-Słowińska D, Hypomagnesemia is underestimated in children with HNF1B mutations: Pediatr Nephrol, 2020; 35(10); 1877-86
14.. Ferrè S, Igarashi P, New insights into the role of HNF-1β in kidney (patho) physiology: Pediatr Nephrol, 2019; 34(8); 1325-35
15.. Okorn C, Goertz A, Vester U, HNF1B nephropathy has a slow-progressive phenotype in childhood – with the exception of very early onset cases: Results of the German Multicenter HNF1B Childhood Registry: Pediatr Nephrol, 2019; 34(6); 1065-75
16.. Duval H, Michel-Calemard L, Gonzales M, Fetal anomalies associated with HNF1Bmutations: Report of 20 autopsy cases: Prenat Diagn, 2016; 36(8); 744-51
17.. Clissold R, Fulford J, Hudson M, Exocrine pancreatic dysfunction is common in hepatocyte nuclear factor 1β-associated renal disease and can be symptomatic: Clin Kidney J, 2018; 11(4); 453-58
18.. Verhave J, Bech A, Wetzels J, Nijenhuis T, Hepatocyte nuclear factor 1β-associated kidney disease: More than renal cysts and diabetes: J Am Soc Nephrol, 2015; 27(2); 345-53
19.. Raaijmakers A, Corveleyn A, Devriendt K, Criteria for HNF1B analysis in patients with congenital abnormalities of kidney and urinary tract: Nephrol Dial Transpl, 2014; 30(5); 835-42
20.. McDonald TJ, Colclough K, Brown R, Islet autoantibodies can discriminate maturity-onset diabetes of the young (MODY) from type 1 diabetes: Diabet Med, 2011; 28(9); 1028-33
21.. Mather H, Nisbet J, Burton G, Hypomagnesaemia in diabetes: Clin Chim Acta, 1979; 95(2); 235-42
22.. Pham PC, Pham PM, Pham SV, Hypomagnesemia in patients with type 2 diabetes: Clin J Am Soc Nephrol, 2007; 2(2); 366-73
23.. Kurstjens S, de Baaij J, Bouras H, Determinants of hypomagnesemia in patients with type 2 diabetes mellitus: Eur J Endocrinol, 2017; 176(1); 11-19
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report  22,759,844
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133