04 June 2021: Articles 
Xanthomatous Inflammatory Infiltrate Involving the Spleen: An Unusual Presentation of Erdheim-Chester Disease and Review of the Literature
Rare disease
Gioia Di Stefano12ABDEF*, Massimo Granai3BCDEF, Francesco Giudici4B, Giuliana Roselli5BCE, Stefano Lazzi1AE, Raffaella Santi2ABDEFDOI: 10.12659/AJCR.931060
Am J Case Rep 2021; 22:e931060






Abstract
BACKGROUND: Erdheim-Chester disease (ECD) is a rare form of non-Langerhans cell histiocytosis characterized by foamy histiocytes, Touton-like giant cells, and fibrosis, typically affecting the diaphyseal and metaphyseal region of the long bones but that can involve any organ or tissue. ECD is usually associated with the BRAF V600E mutation or with other molecular mutations inserted in the MAPK cascade.
CASE REPORT: We present the case of a 63-year-old man with a previous history of myocardial infarction who underwent an emergency splenectomy for splenic rupture after an accidental fall. Histological examination of the spleen showed a diffuse xanthogranulomatous proliferation (CD68+, CD163+, S100–, CD1a–) with rare Touton-like giant cells in the red pulp. Based on the histologic findings, a diagnosis of ECD was made. However, skeletal involvement and BRAF V600E mutation were not detected.
CONCLUSIONS: Cases of non-Langerhans cell histiocytosis that are histologically consistent with ECD in unusual sites have been increasingly described. There is also anecdotal evidence for cases being associated with mutations besides BRAF V600E or with no genetic alteration and no skeletal involvement. Likewise, the spectrum of clinical and molecular features of ECD can be broader than previously considered. Furthermore, there is evidence that various phases of the disease can show different clinical presentations with distinct prognostic impact, according to the mutational spectrum. Recognizing ECD at an early stage allows more effective patient management, and pathologists and clinicians should be aware of the unusual clinical presentations of this rare condition.
Keywords: Erdheim-Chester Disease, Histiocytosis, Non-Langerhans-Cell, splenic rupture, Fibrosis, Mutation, Spleen
Background
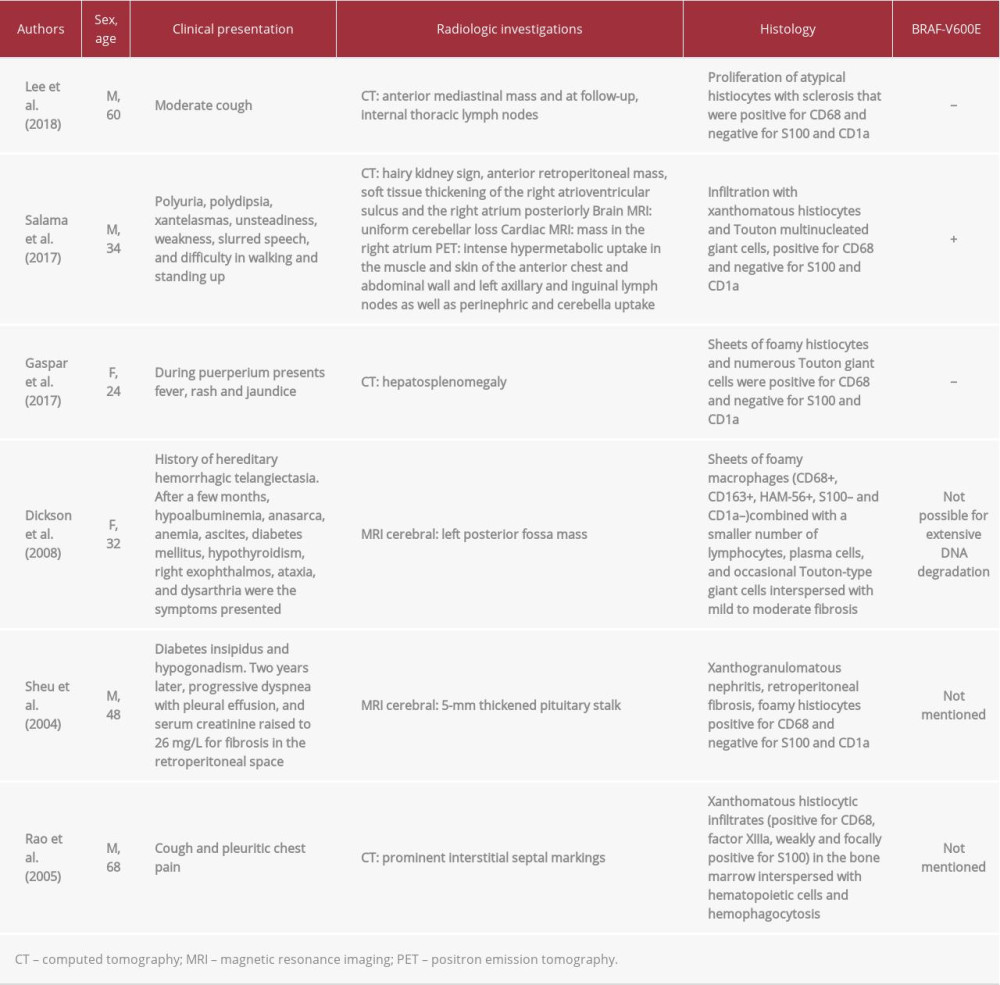
Erdheim-Chester disease (ECD) is a rare form of non-Langer-hans cell histiocytosis with potential systemic involvement. The precise pathogenesis of ECD has not been completely defined. However, the recent discovery of the activation of the RAS-ERK pathway in lesional tissue due to BRAF V600E mutations [1] and other activating mutations involving the mitogen-activated protein kinase (MAPK) pathway has confirmed the neoplastic nature of the disease [2], although cases without mutations have also been identified [3]. Owing to its rarity, the exact prevalence of ECD is unknown; however, roughly 600 cases have been reported in the literature since its first description in 1930 by pathologists Jakob Erdheim (1874–1937) and William Chester (1834–1920) [4]. Diagnosis of ECD is based on histopathologic findings within an appropriate radiological and clinical picture. The consensus diagnostic criteria for ECD require xanthogranulomatous lesions characterized by foamy histiocytes and Touton-like giant cells within a fibrous stroma. Skeletal findings of bilateral and symmetric abnormalities in the diaphyseal and metaphyseal region of the long bones of the legs or, more infrequently, of the arms are typical. Moreover, cases without skeletal involvement have been reported (Table 1). Immunohistochemical staining confirms the non-Langerhans histiocytic lineage of the infiltrate: CD68/CD163-positive with lack of S100 and CD1a/Langerin expression [5]. The presence of xanthogranulomatous lesions is commonly found in several diseases, including infections, lysosomal storage diseases, gamma heavy-chain diseases, lymphomas, and histiocytosis (histiocytosis of the “L” group) [6]. Thus, an accurate anamnestic and clinical evaluation is mandatory to rule out the variety of differential diagnoses.
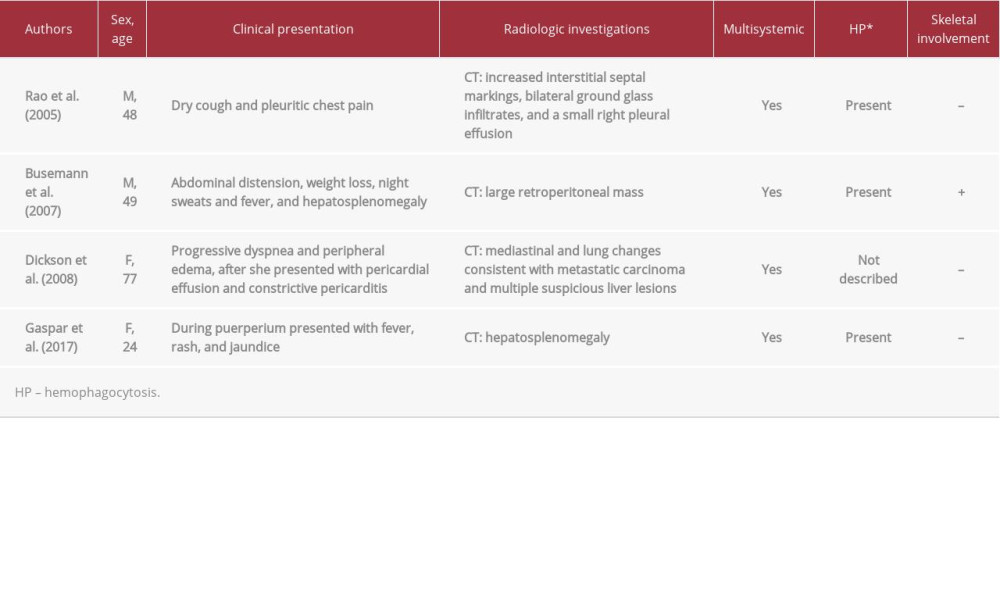
To date, multiple organ involvement has been widely described for ECD, including that of the bones, lungs, skin, retro-orbital tissues, central nervous system, large vessels, kidneys, retroperitoneum, and myocardium [7]. Conversely, evidence of splenic involvement with ECD has been published; however, none of the studies showed splenic involvement as the first manifestation of the disease (Table 2).
We describe a case of xanthomatous inflammatory infiltrate involving the spleen as an unusual presentation of ECD and summarize the features of this extremely infrequent presentation with an additional analysis of the literature.
Case Report
A 63-year-old man was admitted to the emergency department of our hospital with multiple fractures and an abdominal trauma after an accidental fall. The patient’s clinical history revealed hypertension and a myocardial infarction at age 33 years, for which the patient received percutaneous transluminal coronary angioplasty (PTCA) on the left anterior descending coronary artery. Recent echocardiogram results displayed signs of post-myocardial infarction with left ventricular dysfunction (ejection fraction: 40%). A chest computed tomography (CT) scan demonstrated nonspecific peri-bronchial thickening and focal areas of parenchymal consolidation involving the apical segment of the lower lobes and anterior segment of the right superior lobe and enlarged mediastinal lymph nodes (Figure 1A). A CT scan of the abdomen revealed a splenic rupture, and the patient underwent an emergency splenectomy (Figure 1B, 1C). A noncontrast brain CT scan showed cortico-subcortical chronic infarction of the left frontal lobe (Figure 2). At gross examination, the spleen measured 21×6×5 cm, with a weight of 880 g and multiple lacerations.
Histologically, the splenic parenchyma showed a preserved architecture with hemorrhagic extravasation areas and an expanded red pulp (Figure 3A). In the background of a mixed inflammatory infiltrate composed of plasma cells, small lymphocytes, and eosinophils, the red pulp was replaced by a diffuse proliferation of histiocytes with pale and foamy cytoplasm and minimal nuclear pleomorphism, and was occasionally associated with scattered, multinucleated Touton-like giant cells (Figure 3B). The histiocytes were positive for CD68 (Figure 4A), CD163 (Figure 4B), and fascin and were negative for protein S100 (Figure 4C) and CD1a, with a proliferation index of 20%. Special staining for infectious agents, such as Ziehl-Neelsen, Grocott, and Giemsa for Leishmania, were negative, and PCR testing for mycobacterial DNA was also negative. ALK rearrangement and IgG4 were not identified by immunohistochemical analysis. An accurate clinical assessment excluded other possible causes of a splenic localization of a xanthomagranulomatous infiltrate such as storage diseases, infections, gamma heavy-chain disease, lymphomas, and other non-Langerhans cell histiocytosis. Based on the histological findings, a diagnosis of ECD was proposed. The most common mutations (BRAFV600E, KRAS, NRAS) were investigated, and no mutations were found. Subsequently, a bone X-ray of the long bones, in particular of the lower limbs, was performed and was negative (Figure 1D). At follow-up, the patient was in good health, and a watch-and-wait approach was undertaken as a multi-disciplinary team decision, with agreement from the patient.
Discussion
We presented an exceptional and unusual case of non-Langer-hans cell histiocytosis that was histologically compatible with ECD, with primary and single localization in the spleen with no skeletal involvement and the absence of the BRAF V600E mutation. Previous cases described a non-Langherans cell histiocytosis consistent with ECD in different sites, emphasizing that the most crucial diagnostic hallmark of ECD is probably its histopathological characteristics [8].
ECD with single and primary involvement of the spleen has never been described, and the majority of published reports of anecdotal evidence were autoptic findings associated with multisystemic presentations (Table 2). Another exceptional feature of our case was the lack of skeletal involvement, as only 4% of patients with ECD do not present these radiological findings (Table 1).
Previous evidence of ECD with no mutations has been reported [3]. Moreover, it has been demonstrated that mutations of the MAPK cascade are a common finding in advanced phases [9], suggesting that the absence of mutations might represent an early stage of the disease, as in our case. Additionally, associations have been shown to exist between ECD and Langerhans cell histiocytosis and carcinomas, such as papillary thyroid carcinoma, non-small cell lung carcinoma, and colonic adenocarcinoma. The coexistence of Langerhans cell histiocytosis, ECD, and papillary thyroid carcinoma has been found with concurrent BRAF mutation [10,11]. For these reasons, the possibility of underlying neoplasia in the presence of other mutations is an aspect that should be considered when a xanthogranulomatous infiltrate that resembles a non-Lang-erhans histiocytosis is encountered.
Conclusions
Our case is a further example of the diversity and complexity of ECD, as it represents a unique report of the early stage of the disease with merely splenic involvement. Pathologists should be aware of the ECD diagnosis based on histopatho-logical findings, even in cases that do not present the typical clinical-radiological and mutational features.
Figures
References:
1.. Haroche J, Charlotte F, Arnoud L, High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langherans cell histiocytosis: Blood, 2012; 120(13); 2700-3
2.. : WHO classification of the tumors of the haematopoietic and lymphoid tissues, 2017, Lyon, International Agency for Research on Cancer
3.. Ozkaya N, Rosenblum MK, Durham BH, The histopathology of Erdheim-Chester disease: A comprehensive review of a molecularly characterized cohort: Mod Pathol, 2018; 31(4); 581-97
4.. Chester W, Uber lipoidgranulomatose: Virchows Arch Pathol Anat, 1930; 279; 561-602 [in German]
5.. Diamond EL, Dagna L, Hyman DM, Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease: Blood, 2014; 124(4); 483-92
6.. Emile JF, Abla O, Fraitag F, Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineage: Blood, 2016; 127(22); 2672-81
7.. Starkebaum G, Hendrie P, Erdheim-Chester disease: Best Pract Res Clin Rheumatol, 2020; 34(4); 101510
8.. Lee K, Hyeong RK, Roh J, Erdheim-Chester disease presenting as an anterior mediastinal tumor without skeletal involvement: Korean J thorac Cardiovasc Surg, 2018; 51(3); 223-26
9.. Emile JF, Diamond EL, Hélias-Rodzewicz Z, Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease: Blood, 2014; 124(22); 3016-19
10.. Wake L, Xi L, Raffeld M, Jaffe ES, Langerhans cell histiocytosis, non-lang-erhans histiocytosis and concurrent papillary thyroid carcinoma with BRAF V600E mutations: A case report and literature review: Hum Pathol, 2019; 17; 200302
11.. Johnson WT, Patel P, Hernandez A, Langerhans cell histiocytosis and Erdheim-Chester disease, both with cutaneous presentations, and papillary thyroid carcinoma all harboring the BRAF (V600E) mutation: J Cutan Pathol, 2016; 43(3); 270-75
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133