10 October 2021: Articles 
Treatment-Refractory, Primary Immune Thrombocytopenic Purpura in a Patient with Celiac Disease
Unusual clinical course, Challenging differential diagnosis, Unusual or unexpected effect of treatment, Educational Purpose (only if useful for a systematic review or synthesis), Rare coexistence of disease or pathology
Beka Aroshidze1ABCDEFG*, Sos Nalghranyan2ABCDEFG, Gregory Gotlieb2CDF, Burak Erdinc1ABCDEFG, Alok Aggarwal3ABCDEFG, Cherif El Younis4ABCDEF, Boris Avezbakiyav2ABCDEFGDOI: 10.12659/AJCR.931877
Am J Case Rep 2021; 22:e931877






Abstract
BACKGROUND: Immune thrombocytopenic purpura (ITP) is primarily caused by antibody-mediated destruction of platelets. Alterations in immune homeostasis can induce loss of peripheral tolerance and promote the development of self-reactive antibodies. Primary ITP is the diagnosis of exclusion made after the extensive work-up rules out other possible causes of thrombocytopenia. The association between the ITP and other autoimmune disorders is well-established. In recent years, increasing attention has been directed toward the association between celiac disease (CD) and ITP.
CASE REPORT: A 27-year-old man with a history of primary ITP presented with an occasional nosebleed, 1 episode of rectal bleeding, and easy bruising. The patient was later found to have high titers of TTG-IGA and endomysial IGA levels consistent with CD. Our patient not only failed to improve with the gluten-free diet, but also failed multiple lines of treatment including steroids, IVIG, rituximab, eltrombopag, and even a non-traditional treatment for ITP (azathioprine and plasma exchange). The patient’s CD-related antibody titers remained elevated.
CONCLUSIONS: It is possible that in certain cases the alteration of immune response caused by CD with a concurrent elevation of CD-related antibodies can make ITP refractory to all medical management. Whether or not this refractoriness to treatment is related to the persistently elevated antibody titers of CD or unknown genetic relationship between ITP and CD remains not entirely clear and warrants further molecular, immunologic, and genetic analysis.
Keywords: Allergy and Immunology, Hematologic Diseases, Thrombocytopenia, celiac disease, Diet, Gluten-Free, Humans, Plasma Exchange, Purpura, Thrombocytopenic, Idiopathic, rituximab
Background
Immune thrombocytopenic purpura (ITP) is an incompletely understood disease, likely related to antibody-mediated destruction of platelets (typically through IgG-antibodies directed against GpIIb/IIIa on the platelet membrane). Other mechanisms are likely to be important, including autoreactive Tc cells, as well as humoral and cellular autoimmunity directed at megakaryocytes, causing impaired platelet production.
Alterations in immune homeostasis might induce loss of peripheral tolerance and promote the development of self-reactive antibodies [1]. The association between the ITP and other autoimmune disorders (including systemic lupus erythematosus, antiphospholipid syndrome, autoimmune lymphoproliferative syndrome) is well-established. In recent years, increasing attention has been directed toward the association between ITP and celiac disease (CD) [2].
CD is a common cause of intestinal malabsorption secondary to ingestion of gluten-containing proteins in genetically predis-posed individuals. This disease affects the small bowel through the hypersensitivity reaction that causes inflammation of the mucosa, hyperplasia of the crypts, and atrophy of the villi. A gluten-free diet is the most important step in management. CD is associated with multiple autoimmune diseases (eg, type 1 diabetes, autoimmune thyroid disease, and atopic dermatitis).
Co-occurrences of CD and ITP are reported; however, these patients are usually successfully treated with the gluten-free diet and/or medical management (eg, steroids).
In the current case report, we discuss a case of primary ITP associated with CD, which turned out to be refractory to almost all known treatments of ITP, with some improvement of platelet count after surgical management (splenectomy).
Case Report
A 27-year-old man was referred from the primary care physician’s office to the emergency department for a low platelet count of 4×103/uL. The patient experienced bleeding from his nose 2 weeks prior, 1 episode of rectal bleeding, and chronic frequent petechiae on the skin. In addition, the patient had prolonged bleeding from minor injuries (up to 2–3 hours). Interestingly, the other presenting sign was hair loss on the scalp (Figure 1).
The patient was not on any medication and had not received any vaccinations for the past years. His past medical history included similar minor bleeding from thrombocytopenia diagnosed as primary immune thrombocytopenic purpura (ITP) in Uzbekistan in 2017 (2 years before migration to the USA). He had completed several courses of steroids and IVIG in Uzbekistan and India, with a suboptimal response. In India, the patient had refused to have a splenectomy. The patient also had a distant past medical history of tuberculosis of the right femur treated medically and surgically in Uzbekistan.
Except for steroids and IVIG, the patient had not received any other treatments for the last 5 years. All medications that the patient received after the initial presentation are described below.
On presentation, the patient did not have any major bleeding or other active concerns. He had asymptomatic anemia (hemoglobin level was 11.6 g/dL) with low ferritin (21 ng/mL on the first admission, subsequently decreased to 14 ng/mL) and mild microcytosis (MCV 75–79 fL). Mean platelet volume was borderline high or elevated (as high as 13.7). Vitamin B12, fo-late, coagulation studies (PT, INR, PTT), and thyroid function were within normal range.
A neurological exam was unremarkable. He did not have any evidence of arthritis/arthralgia, fever, chills, night sweats, or weight changes. ESR was normal (10 mm), and ANA and rheumatoid factor results were negative. His kidney function remained normal during the entire treatment course (creatinine of 0.62–0.81 mg/dL). ANCA/complement and urine protein levels were not obtained because of the low suspicion of vasculitides and other rheumatologic/connective tissue diseases. The patient did not experience any signs or symptoms of thrombosis.
Laboratory test results for HIV, HBV, HCV, ANA, RF, RPR were all negative, and there was no evidence of EBV, CMV, VZV, or rubella. CXR ruled out active pulmonary TB. X-rays of the right femur, right knee, and right tibia/fibula were negative for acute changes. Computed tomography of the chest, abdomen, and pelvis with i.v. contrast revealed no hepatosplenomegaly and no lymph node enlargement. Peripheral smear showed the absence of giant platelets, occasional teardrop cells, and normal morphology of WBCs.
Bone marrow biopsy (Figure 2) revealed slightly hypercellular marrow (70%) with trilineage hematopoiesis. The myeloid-toerythroid ratio (M: E) appeared within normal range; increased numbers of megakaryocytes were present, consistent with immune thrombocytopenic purpura (ITP); no evidence of lymphoma or leukemia was seen; no stainable storage iron was noted; and no marrow fibrosis or granulomatous inflammation/ necrosis were identified. Given the past medical history of TB, the possibility of disseminated TB with bone marrow involvement was ruled out by negative AFB stain and culture for AFB.
After the described extensive evaluation ruled out other causes or triggers, the diagnosis of primary ITP was made. Hereditary megakaryocyte dysfunction was unlikely, as these are rare disorders usually characterized by specific clinical findings (eg, short stature and arm developmental disorders in Fanconi anemia and Diamond-Blackfan anemia), earlier onset (often in infancy, eg, in congenital amegakaryocytic thrombocytopenia [C-MPL mutation]), involvement of other cell lineages, peripheral smear, and bone marrow changes (eg, large platelets with reduced α-granules and the development of myelofibrosis inside the bone marrow with Gray platelet syndrome [NBEAL2 mutation]) and some family history. Our patient, with no significant family history, developed petechiae and frequent nose-bleeds in his 20s; he was subsequently found to have isolated thrombocytopenia, and had normal physical exam without specific clinical findings, peripheral smear, and bone marrow biopsy, compatible with the diagnosis of primary ITP. Bone marrow demonstrating an increased number of megakaryocytes and no iron storage is not consistent with hereditary thrombocytopenia, but rather with peripheral destruction of platelets characteristic for ITP.
Treatment with prednisone (1 mg/kg daily) and rifampin (as prophylaxis against the reactivation of TB), and IVIG (1 g/kg) were started. Platelet counts slightly improved (from 10 to 15×103/uL) and the patient was discharged with outpatient weekly rituximab treatment (along with steroids). Rituximab was given in a total of 3 cycles; nevertheless, the patient visited the ED 2 times while on rituximab treatment, with severe persistent thrombocytopenia and petechiae on the left arm. In the ED, platelet transfusion, IVIG, and IV iron were given. The patient was upset about the ineffectiveness of the treatment and refused to be admitted.
Eltrombopag was started (initially 50 mg daily, subsequently increased to TID dosing), and 2 more doses of IVIG were administered. His platelet count remained at 4×103/uL.
Because there were no significant bleeding episodes reported that would explain the complete absence of stainable iron on bone marrow biopsy, an iron deficiency work-up was performed. The patient was found to have a high celiac disease (CD) antibody panel result: elevated titers of tTG-IgA (>100 U/mL) and endomysial IGA levels (1: 80). When specifically asked, the patient stated he ate a gluten-free diet because the ingestion of gluten-containing products was generally resulting in abdominal pain and diarrhea.
Capsule endoscopy was preferred over the EGD (given the patient’s low platelet count), and the findings were consistent with CD (Figure 3). Genetic testing for HLA-DQ2 or DQ8 haplotypes was not obtained because this test is useful only in ruling out CD (negative predictive value of >99%, positive predictive value of 12%). The confirmatory criterion standard diagnostic test would be a duodenal biopsy; however, it was not done because of the high risk of bleeding in a patient with persistently low platelet counts.
The diagnosis of CD was based on: 1) Positive CD-related antibodies; 2) The original resolution of GI symptoms with the gluten-free diet; 3) Villous atrophy observed macroscopically on the endoscopy.
A PET scan was performed, which excluded any extra-splenic manifestation, including indolent B cell lymphoma in the gastrointestinal tract that could have been the reason for refractory ITP on top of his CD (Figure 4).
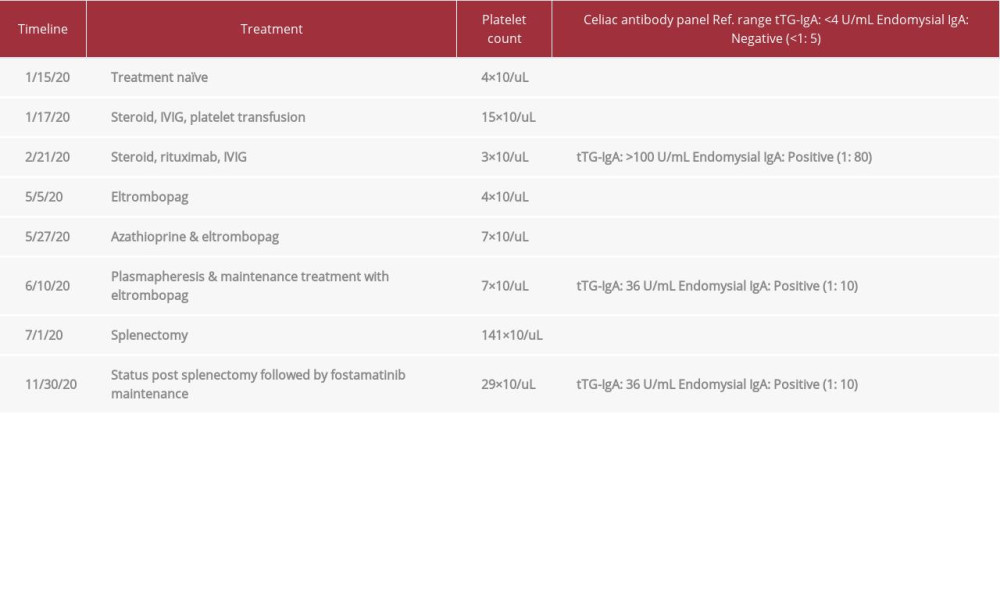
The patient was started on azathioprine (100 mg daily) to decrease the CD-related antibody titers. However, there was no improvement in platelet count. He received plasmapheresis to remove CD-related antibodies and potentate the response to anti-ITP medications: 3 L of plasma was successfully exchanged, with 2 L of albumin 5% and 1 L of FFP. Azathioprine 50 mg BID and eltrombopag 50 mg TID regimen was continued and ITP treatment options were re-challenged. However, the platelet count remained below 10×103/uL. Although CD-specific antibody titer was decreasing (Table 1), ITP remained resistant to treatment.
Fostamatinib (100 mg BID) was started as a last resort before performing a splenectomy. However, in the setting of persistent chronic severe thrombocytopenia, and worsening worrisome symptoms of diffuse petechiae (especially over the torso and arms), laparoscopic splenectomy was performed without complications along with the platelet transfusion.
Postoperative spleen pathology showed splenic parenchyma exhibiting reactive follicles with well-developed germinal centers with neutrophils, histiocytes, and hemorrhage, which are changes consistent with ITP. No evidence of lymphoma was seen.
After the surgery, the platelet count improved to 141×103/uL, but during the next few months, it dropped to 29×103/uL. The patient’s CD-related antibody titers remained elevated. The patient did not have any symptoms whatsoever.
Discussion
Primary ITP is a diagnosis of exclusion. After an extensive evaluation rules out other causes or triggers, the diagnosis of primary ITP is made. In our case, history, physical exam, laboratory work-up, peripheral smear, and bone marrow biopsy ruled out the other possible etiologies of thrombocytopenia.
The patient received only the medications listed in the present case report.
The first case report of the coexistence of immune thrombocytopenic purpura (ITP) and celiac disease (CD) in a child was described in 1988 [3]. Since then, co-occurrences of CD and ITP have been widely described in the literature, which suggests that these 2 conditions are associated and are not merely coincidental. Moreover, it was found that both diseases may have similar autoimmune mechanisms [4]. The innate immune system is essential in the pathogenesis of CD, and Toll-like receptors (TLRs) are key players in the innate immune system [5]. Zanoni et al [6] reported that patients with CD have a subset of transglutaminase antibodies that activate TLR4. In addition, it is thought that the TLR4 expression in platelets is a prerequisite for thrombocytopenia [7]. Eliakim et al suggested that the link between CD and ITP may be genetic through the human leukocyte antigen (HLA) system [8].
CD is known to be associated with alopecia areata (AA), which is a polygenic, multifactorial pathology that can present with multiple patterns of non-scarring hair loss. A biopsy of the damaged skin revealing lymphocytic infiltrate (localized usually around the bulb or in the lower part of the hair follicle) suggests a possible autoimmune etiology of AA. The existence of organ-specific autoantibodies has been shown for both CD and AA [9], as well as infiltration of the lesion site by T lymphocytes [10]. Moreover, some studies have focused on chromosome 6 (specifically on the HLA region) as the most likely location for the genes that result in susceptibility to AA [11]. AA seen in our patient may be the presentation of immuno-logically active celiac disease.
We discussed the case of primary ITP associated with CD, which is special because of the multiple treatment modalities used while treating the patient’s condition and the unusual refractoriness of the ITP. In most of the available case reports, these patients respond well to a gluten-free diet [12,13]. There have been exceptions to this, such as Stenhammar and Ljunggren’s case report [3]. Our patient not only failed to improve with the gluten-free diet, but also failed multiple lines of treatment, including steroids, IVIG, rituximab, eltrombopag, and even a non-traditional treatment for ITP that involved azathioprine and plasma exchange (Figure 5). For the last 6 months, his platelet count varied from 3×103/uL to 15×103/uL, despite medical management and multiple supportive transfusions. After the surgery, the platelet count improved to 141×103/uL, but during the next few months, it dropped to 29×103/uL. The patient’s CD-related antibody titers remained elevated, but he remained asymptomatic.
Conclusions
It is reasonable to think that the refractoriness of primary immune thrombocytopenic purpura (ITP) in certain cases arises from the altered immune response caused by celiac disease (CD). The consistent elevation in CD-related antibody titers might be leading to the failure of all known medical interventions to treat ITP. This case report shows that the management of coexistent ITP and CD can become challenging, and warrants further molecular, immunologic, and genetic analysis of these 2 diseases.
Figures
References:
1.. George JN, Arnold DM, Immune thrombocytopenia (ITP) in adults: Second-line and subsequent therapies – UpToDate, 2020; 1-36 In: UpToDate.
2.. Bibbò S, Fozza C, Pes GM, Rojas R, Increased frequency of immune thrombocytopenic purpura in coeliac disease and vice versa: A prospective observational study: Gastroenterol Res Pract, 2018; 2018; 4138434
3.. Stenhammar L, Ljunggren CG, Thrombocytopenic purpura and coeliac disease: Acta Paediatr Scand, 1988; 77(5); 764-66
4.. Olén O, Montgomery SM, Elinder G, Increased risk of immune thrombocytopenic purpura among inpatients with coeliac disease: Scand J Gastroenterol, 2008; 43(4); 416-22
5.. Stepniak D, Koning F, Celiac disease-sandwiched between innate and adaptive immunity: Hum Immunol, 2006; 67(6); 460-68
6.. Zanoni G, Navone R, Lunardi C, In celiac disease, a subset of autoantibodies against transglutaminase binds toll-like receptor 4 and induces activation of monocytes: PLoS Med, 2006; 3(9); e358
7.. Aslam R, Speck ER, Kim M, Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo: Blood, 2006; 107(2); 637-41
8.. Eliakim R, Heyman S, Kornberg A, Celiac disease and keratoconjunctivitis: occurrence with thrombocytopenic purpura: Arch Intern Med, 1982; 142(5); 1037
9.. Pratt CH, King LE, Messenger AG, Alopecia areata: Nat Rev Dis Prim, 2017; 3; 17011
10.. Carroll JM, McElwee KJ, King LE, Gene array profiling and immuno-modulation studies define a cell-mediated immune response underlying the pathogenesis of alopecia areata in a mouse model and humans: J Invest Dermatol, 2002; 119(2); 392-402
11.. Villasante Fricke AC, Miteva M, Epidemiology and burden of alopecia area-ta: A systematic review: Clin Cosmet Investig Dermatol, 2015; 8; 397-403
12.. Dogan M, Sal E, Akbayram S, Concurrent celiac disease, idiopathic thrombocytopenic purpura and autoimmune thyroiditis: A case report: Clin Appl Thromb, 2011; 17(6); E13-16
13.. Sarbay H, Cosan Sarbay B, Akın M, Celiac disease presenting with immune thrombocytopenic purpura: Case Rep Hematol, 2017; 2017; 6341321
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133