20 July 2021: Articles 
A Report of 2 Infant Siblings with Progressive Intrahepatic Familial Cholestasis Type 1 and a Novel Homozygous Mutation in the ATP8B1 Gene Treated with Partial External Biliary Diversion and Liver Transplant
Unusual clinical course, Unusual or unexpected effect of treatment, Rare disease, Congenital defects / diseases
Irena Jankowska1ABCDEF, Joanna Pawłowska1ACDE, Marek Szymczak2BDE, Hor Ismail2BDE, Dorota Broniszczak2BDE, Joanna Cielecka-Kuszyk3BCE, Piotr Socha1ACDE, Dorota Jarzębicka1BDEF*, Piotr Czubkowski1ABCDEFDOI: 10.12659/AJCR.932374
Am J Case Rep 2021; 22:e932374






Abstract
BACKGROUND: Current treatment options for progressive intrahepatic familial cholestasis type 1 (PFIC-1) comprise ursodeoxycholic acid (UDCA), partial external biliary diversion (PEBD), and liver transplantation (LTx). The role and timing of LTx in PFIC-1 remains debated. We present 2 case reports of male siblings with PFIC-1 who benefited from different treatments.
CASE REPORT: Both siblings harbored a homozygous truncating mutation in ATP8B1 characteristic for PFIC-1 and both underwent PEBD after unsuccessful UDCA treatment at the age of 7 and 5 months, respectively. The older brother, after initial improvement of symptoms, developed severe pruritus, cholestasis, and diarrhea 9 months after PEBD and underwent LTx at the age of 16 months. Chronic diarrhea and abnormal transaminases activity appeared soon after transplantation. A liver biopsy was performed 3 months after LTx and showed severe macrovesicular steatosis (95%). Sixteen months after LTx, total biliary diversion was performed, with rapid relief from diarrhea and significant regression of graft steatosis by <30%. In his brother we observed persistent severe pruritus and cholestasis after PEBD, but we decided to postpone LTx due to lack of a living related donor and risk of graft steatosis. Eight months after PEBD, bilirubin and bile acids significantly decreased and pruritus disappeared completely. Currently, in 5-year follow-up, liver function is stable and he has no pruritus.
CONCLUSIONS: The good effect of PEBD may be delayed in PFIC-1, even in severe mutation; thus, the decision to perform LTx should be made cautiously. Total biliary diversion is an efficient procedure in case of persistent symptoms after LTx and can reverse graft steatosis in children with PFIC-1.
Keywords: Biliary Tract Surgical Procedures, Cholestasis, Transplants, Adenosine Triphosphatases, Child, Cholestasis, Intrahepatic, Infant, Liver Transplantation, Mutation, Siblings
Background
Progressive familial intrahepatic cholestasis type 1 (PFIC-1) is an autosomal recessive disorder caused by biallelic pathogenic variants in the
Case Reports
CASE 1:
A 6-week-old boy was admitted to our hospital after an episode of prolonged subcutaneous bleeding after vaccination. He was born at term with weight of 2920 g, of healthy, unrelated parents. It was a second pregnancy after previous miscarriage at the 6th week of pregnancy. There was no family history of liver disease. At admission, the patient was jaundiced with subcutaneous hematoma at the site of vaccination. Spleen and liver were not enlarged and stools were yellow. Laboratory tests showed severe coagulopathy (INR was undetectable) and cholestasis with normal GGTP (Table 1). Vitamin K and blood plasma were transfused; UDCA (20 mg/kg/day) and vitamin supplementation (A, D, E, K) were started. After excluding other causes of cholestasis, PFIC-1 was confirmed genetically. Within the next months, the patient presented with poor growth and developed severe pruritus with rapid increase of serum bilirubin and bile acids. At the age of 7 months, partial external biliary diversion (PEBD) was performed. The liver biopsy showed changes consistent with PFIC1, with bland cholestasis, hepatocytic rosette formation around bile plugs, fibrosis, and normal bile ducts without ductular proliferation. After the initial disappearance of pruritus and normalization of serum bile acids, the cholestasis aggravated, and at the age of 16 months (8 months after PEBD), the patient underwent living related liver transplantation from his mother. Concurrently, the external stoma was removed. No surgical complications occurred in the post-transplant period, and standard immunosuppression with tacrolimus and mycofenolate mofetil (MMF) was started. Severe chronic diarrhea (5–7 liquid stools per day) appeared 1 month after transplantation. Repeated stool cultures and parasite examination were negative and there was no response to cholestyramine and MMF discontinuation (prednisone was introduced). Three months after LTx, due to abnormal transaminases activity, a liver biopsy was performed, which showed diffuse macrovesicular steatosis (95%) with no other changes (Figure 1A). Due to unremitting diarrhea and growth retardation 16 months after transplantation, total external biliary diversion (EBD) was performed a with previously described technique [8]. The distal part of the Roux loop was disconnected from the bowel tract and opened as a terminal jejunostomy. Liver biopsy at the time of EBD showed severe steatosis and moderate fibrosis (Figure 1B). After the surgery, rapid relief from diarrhea was observed and follow-up liver biopsies showed significant regression of steatosis and fibrosis (Figure 1C, 1D).
Five years after LTx, the patient has no diarrhea and his weight is between the 25th and 50th percentiles and his height is between the 10th and 25th percentiles. A clinical summary is presented in Table 1.
CASE 2:
The younger brother who was born at term after uncomplicated pregnancy developed cholestasis within the first 4 weeks after birth. Treatment with UDCA (20 mg/kg/day) was ineffective and during the next months the patient developed severe pruritus and coagulopathy (Table 2). PEBD was performed at the age of 5 months. Microscopic examination of the liver biopsy specimen revealed distortion of the normal architecture with severe fibrosis, giant cell transformation of the liver cells without steatosis, severe cholestasis, and rosette formation around the bile plugs, with non-specific mild inflammation. After initial transient improvement, the pruritus recurred and cholestatic parameters increased. Due to the lack of a living donor and stable liver function parameters, we decided to postpone the transplant decision. Meanwhile, the patient remained on UDCA and vitamin supplementation (A, D, E, K). Eight months after PEBD, bilirubin rapidly decreased to 2 mg/dl, bile acids concentration changed to 65 ng/ml, and pruritus disappeared completely. Afterwards, complete normalization of cholestatic parameters was observed. Currently, after 4-year follow-up, the patient presents with normal lab test results, itching occasionally (only when infected or tired), and growing well.
Discussion
We presented 2 case reports of male siblings, sharing the same homozygous mutation in
PFIC-1 is an inherited disorder leading to progressive liver damage, severe pruritus, and diarrhea, significantly influencing survival and quality of life [1,2]. Current medical treatment options are limited to administration of ursodeoxycholic acid, nutrition support with fat-soluble vitamin supplementation, and anti-pruritus agents of varying effectiveness, like rifampicin, cholestyramine or ondansetron [1,2,10–17]. Moreover, neither of the above seems to affect the primary disease process. Over the last decades, surgical techniques causing disruption of the enterohepatic circulation have been employed and they remain the mainstay of primary treatment in PFIC. They include partial or total external biliary diversion, internal biliary diversion, and liver transplantation [6–16,18–19].
Unlike in PFIC-2, LTx has significant limitations in PFIC-1 in which extrahepatic features like diarrhea, failure to thrive, or pancreas dysfunction can aggravate after transplantation. Moreover, patients often develop graft steatosis or steatohepatitis possibly progressing to cirrhosis with need of re-transplantation [3,5–10]. One of the possible responsible mechanisms could be decompensation of malfunctioning
Medical symptomatic treatment options for protracted diarrhea are limited. Some authors described a good effect of bile adsorptive resin therapy, but that was not satisfactory in our patients [4,17]. Several subsequent reports showed outstanding outcomes of post-transplant biliary diversion, with relief of diarrhea and regression of liver steatosis (Table 3). Some authors described good results of internal biliary diversion used preemptively at the time of LT as a stoma-free procedure to prevent postoperative graft steatosis or as a next step after PEBD [9,18].
Bull et al analyzed 42 PFIC-1 patients, and reported no difference in the frequency of clinically important poor outcomes of PEBD between FIC1 and BSEP patients, but found a greater proportion of BSEP-other compared to FIC1 patients who progressed to cirrhosis [10].
Squires et al [19] reported 8 patients with PFIC after PEBD and follow-up after 32 months on average (range, 15–65 months). They observed that total bilirubin levels dropped below 2 mg/ dL in all patients by 8 months after PEBD (itching improved after PEBD within 3 months in all patients). Despite observed overall clinical improvement after PEBD, the authors reported recurrent transient episodes of cholestasis, often in combination with both worsening pruritus and declining vitamin levels.
Our patients harbored the homozygous state of a very rare mutation (2097+2T>C) causing in-frame deletion and subsequent protein truncation. Only 1 case was previously reported with the same mutation, who received cadaveric liver transplant at 5.5 years, followed by diarrhea exacerbation requiring parenteral nutrition, appearance of liver steatosis and no catch-up of stature growth [5] Diarrhea control was gained with cholestyramine. Ten years after LTx, there is persistent graft steatosis, slight lobular fibrosis, and portal fibrosis. Heterozygous variants of this mutation were reported twice previously and tend to have milder phenotype not leading to liver transplantation [1,19].
The delayed and sustained effect of PEBD observed in our patient with PFIC-1 is unclear. One of the possible explanations may be some interplay among overall bile acid pool, FXR expression, function of ASBT (apical sodium-dependent bile acid transporter), and microbiota alterations. Hypothetically, cholestasis develops presumably because of both enhanced ileal uptake of bile salts via upregulation of the apical sodium-dependent bile acid transporter and diminished canalicular secretion of bile salts secondary to downregulation of the bile salt excretory pump [2,20]. How biliary diversion affects these pathways could be a key to development of targeted medical treatment.
Our findings should be interpreted with caution due to some methodological limitations resulting mainly from retrospective chart review.
Conclusions
In conclusion, a good effect of PEBD may occur with delay in PFIC-1, even in severe mutations; thus, the decision to perform LTx should be made cautiously. Post-transplant diarrhea and graft steatosis are reversible by biliary diversion. A combination of concurrent LTx and biliary diversion may be considered individually in PFIC-1 patients.
References:
1.. Bull LN, van Eijk MJ, Pawlikowska L, A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis: Nat Genet, 1998; 18; 219-24
2.. Bull LN, Thompson RJ, Progressive familial intrahepatic cholestasis: Clin Liver Dis, 2018; 22; 657-69
3.. Pawlikowska L, Strautnieks S, Jankowska I, Differences in presentation and progression between severe FIC1 and BSEP deficiencies: J Hepatol, 2010; 53; 170-78
4.. Davit-Spraul A, Fabre M, Branchereau S, ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): Phenotypic differences between PFIC1 and PFIC2 and natural history: Hepatology, 2010; 51; 1645-55
5.. Lykavieris P, van Mil S, Cresteil D, Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: No catch-up of stature growth, exacerbation of diarrhoea, and appearance of liver steatosis after liver transplantation: J Hepatol, 2003; 39; 447-52
6.. Miyagawa-Hayashino A, Egawa H, Yorifuji T, Allograft steatohepatitis in progressive familial intrahepatic cholestasis type 1 after living donor liver transplantation: Liver Transpl, 2009; 15; 610-18
7.. Usui M, Isaji S, Das BC, Liver retransplantation with external biliary diversion for progressive familial intrahepatic cholestasis type 1: A case report: Pediatr Transplant, 2009; 13; 611-14
8.. Nicastro E, Stephenne X, Smets F, Recovery of graft steatosis and protein-losing enteropathy after biliary diversion in a PFIC 1 liver transplanted child: Pediatr Transplant, 2012; 16; E177-82
9.. Alrabadi LS, Morotti RA, Valentino PL, Biliary drainage as treatment for allograft steatosis following liver transplantation for PFIC-1 disease: A single-center experience: Pediatr Transplant, 2018; 22; e13184
10.. Bull LN, Pawlikowska L, Strautnieks S, Outcomes of surgical management of familial intrahepatic cholestasis 1 and bile salt export protein deficiencies: Hepatol Commun, 2018; 30(2); 515-28
11.. Kaliciński PJ, Ismail H, Jankowska I, Surgical treatment of progressive familial intrahepatic cholestasis: Comparison of partial external biliary diversion and ileal bypass: Eur J Pediatr Surg, 2003; 13; 307-11
12.. Jankowska I, Czubkowski P, Kaliciński P, Ileal exclusion in children with progressive familial intrahepatic cholestasis: J Pediatr Gastroenterol Nutr, 2014; 58; 92-95
13.. Shneider BL, Progressive intrahepatic cholestasis: Mechanisms, diagnosis and therapy: Pediatr Transplantation, 2004; 8; 609-12
14.. Whitington PF, Whitington GL, Partial external diversion of bile for the treatment of intractable pruritus associated with intrahepatic cholestasis: Gastroenterology, 1988; 95; 130-36
15.. Emond JC, Whitington PF, Selective surgical management of progressive intrahepatic cholestasis (Byler’s disease): J Pediatr Surg, 1995; 30; 1635-41
16.. Lemoine C, Bhardwaj T, Bass LM, Superina RA, Outcomes following partial external biliary diversion in patients with progressive familial intrahepatic cholestasis: J Pediatr Surg, 2017; 52; 268-72
17.. Egawa H, Yorifuji T, Sumazaki R, Intractable diarrhoea after liver transplantation for Byler’s disease: Successful treatment with bile adsorptive resin: Liver Transpl, 2002; 8; 714-16
18.. Mali VP, Fukuda A, Shigeta T, Total internal biliary diversion during liver transplantation for type 1 progressive familial intrahepatic cholestasis: A novel approach: Pediatr Transplant, 2016; 20; 981-86
19.. Squires JE, Celik N, Morris A, Clinical variability after partial external biliary diversion in familial intrahepatic cholestasis 1 deficiency: J Pediatr Gastroenterol Nutr, 2017; 64(3); 425-30
20.. Chen F, Ananthanarayanan M, Emre S, Progressive familial intrahepatic cholestasis, type 1, is associated with decreased farnesoid X receptor activity: Gastroenterology, 2004; 126(3); 756-64
Tables
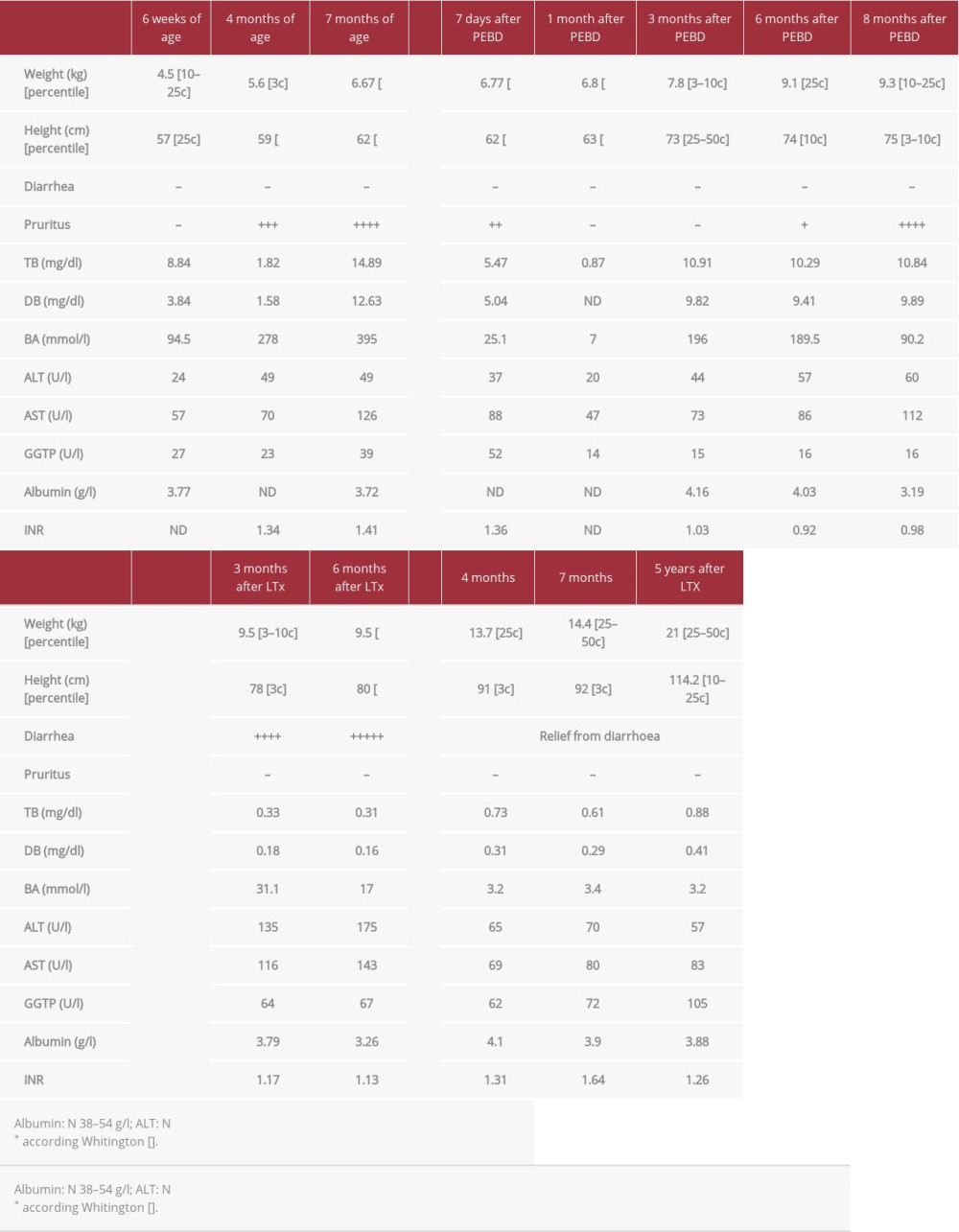 Table 1.. Patient 1. ATP8B1 NM_005603.4: C.2097+2T>C.
Table 1.. Patient 1. ATP8B1 NM_005603.4: C.2097+2T>C.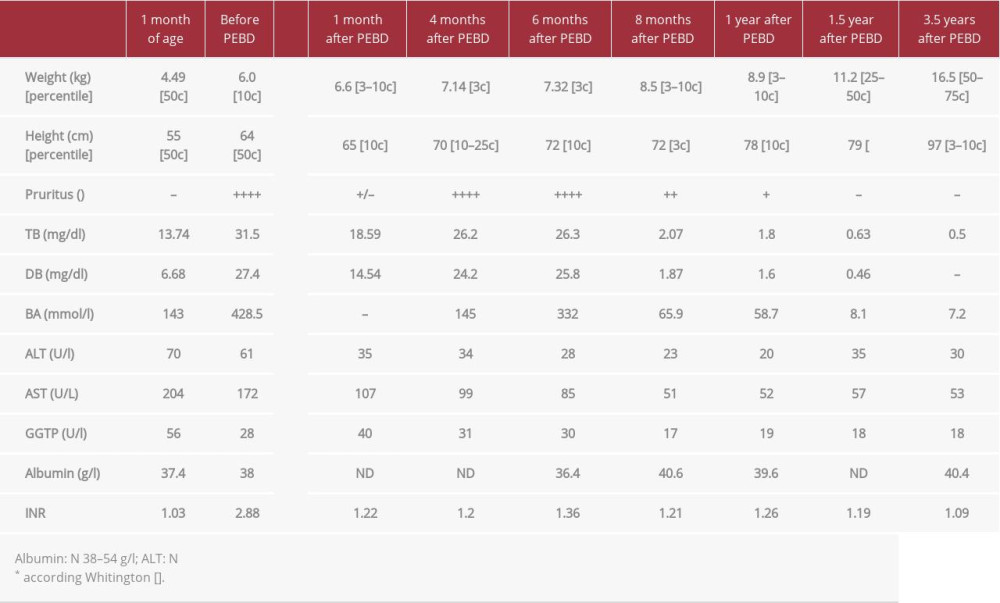 Table 2.. Patient 2. ATP8B1 NM_005603.4: C.2097+2T>C.
Table 2.. Patient 2. ATP8B1 NM_005603.4: C.2097+2T>C.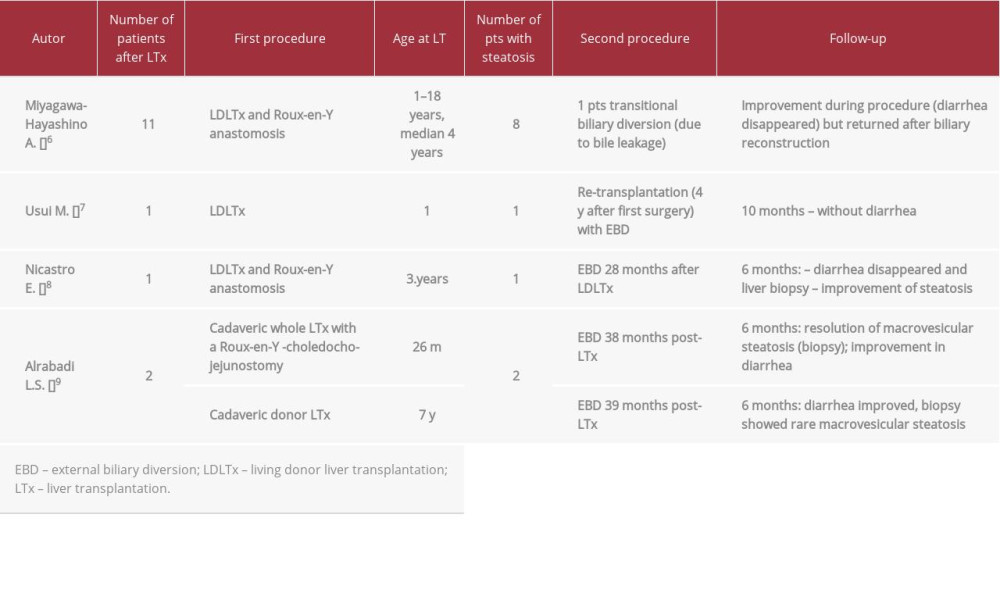 Table 3.. Rescue procedure in PFIC 1 patients with diarrhea/steatosis after LTx.Table 1.. Patient 1. ATP8B1 NM_005603.4: C.2097+2T>C.Table 2.. Patient 2. ATP8B1 NM_005603.4: C.2097+2T>C.Table 3.. Rescue procedure in PFIC 1 patients with diarrhea/steatosis after LTx.
Table 3.. Rescue procedure in PFIC 1 patients with diarrhea/steatosis after LTx.Table 1.. Patient 1. ATP8B1 NM_005603.4: C.2097+2T>C.Table 2.. Patient 2. ATP8B1 NM_005603.4: C.2097+2T>C.Table 3.. Rescue procedure in PFIC 1 patients with diarrhea/steatosis after LTx. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133