08 June 2021: Articles 
First 2 Fabry Cases with Novel Mutation and Their Associated Clusters in Malaysia
Challenging differential diagnosis, Rare disease, Educational Purpose (only if useful for a systematic review or synthesis)
Andy Sing Ong Tang1ABCDEF*, Qi Ying Wong1BCD, Ingrid Pao Lin TingDOI: 10.12659/AJCR.932923
Am J Case Rep 2021; 22:e932923






Abstract
BACKGROUND: No cases of Fabry disease (FD) have been reported thus far in Malaysia. We aimed to report the demographic characteristics, clinical manifestations, molecular results, and treatment outcomes of 2 FD cases. This study was a retrospective review of 2 family clusters of FD on follow-up in Sarawak, Malaysia.
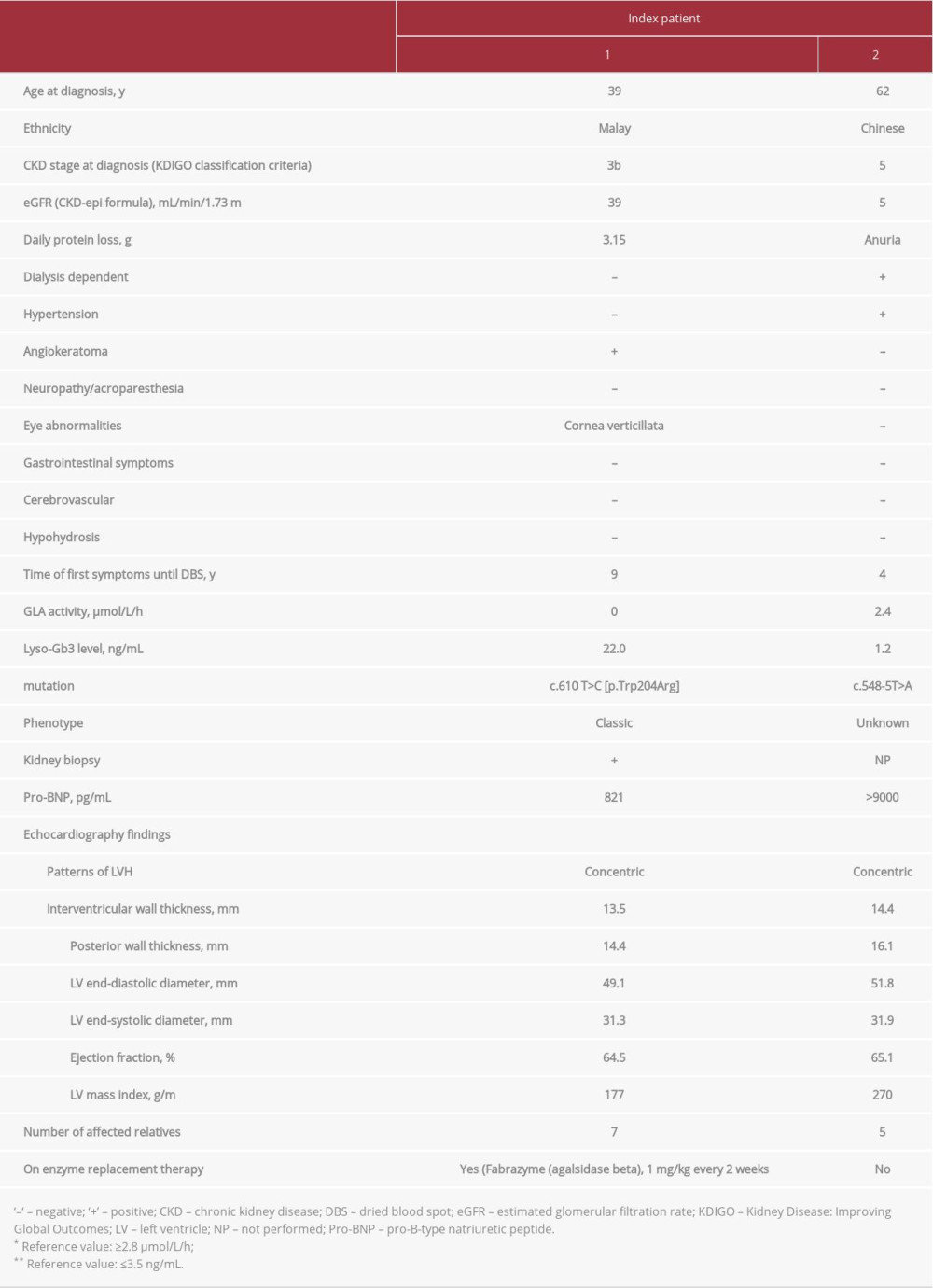
CASE REPORT: Two index patients were confirmed to have FD. Index patient 1, who had nephrotic-range proteinuria and cornea verticillata, carried a variant within exon 4 of the GLA gene: c.610 T>C (p.Trp204Arg). Agalsidase beta (Fabrazyme®) enzyme replacement therapy was initiated, with the absence of neutralizing antibody after 24 months. No hypersensitivity or adverse reactions were reported. The patient’s proteinuria and renal function remained stable. Other family members who carried the same mutation were asymptomatic. Index patient 2, who had residual activity of α-galactosidase A and a normal globotriaosylsphingosine level, carried a novel GLA mutation of c.548-5T>A. He was diagnosed with end-stage renal disease on regular dialysis and had nonspecific headache with 1 episode of seizure a few years prior to FD genetic screening. One brother had chronic neuropathic pain but refused further investigations. Other family members who had the same mutation were asymptomatic. This mutation has never been reported in literature, and its pathogenicity warrants further studies.
CONCLUSIONS: It is of utmost importance to increase awareness of FD among clinicians, so that appropriate screening may be done to determine its true prevalence and prompt treatment can be initiated early.
Keywords: enzyme replacement therapy, Fabry Disease, Malaysia, Mutation, Kidney Failure, Chronic, alpha-Galactosidase
Background
Fabry disease (FD), an X-linked recessive lysosomal storage disorder of glycosphingolipid catabolism, results from deficient or absent activity of the lysosomal enzyme α-galactosidase A (α-gal A). The enzymatic defect leads to progressive accumulation of the glycolipid globotriaosylceramide (GB3 or GL3) and globotriaosylsphingosine (lyso-GL3 or lyso-Gb3) in the lysosomes in the cells of most organs, leading to selective multi-organ damage affecting the kidneys, heart, nervous system, eyes, vascular, and other systems [1,2].
The incidence of FD varies widely in different countries, and it can present in many variant forms. In some countries such as Taiwan, FD is part of a universal newborn screening program [3]. In contrast, Malaysia has restricted diagnostic kits and limited accessibility to treatment. To our knowledge, no cases of FD have been reported thus far in Malaysia. We report 2 cases of FD in Sarawak, Malaysia, along with their demographic characteristics, clinical manifestations, treatment options, and outcomes. We also aimed to identify family clusters through enzyme testing and genetic screening. We hope to increase awareness of the disease, so that appropriate screening may be done to determine its true prevalence and appropriate treatment can be initiated early.
This study was a retrospective review of 2 cases of FD diagnosed in Miri Hospital, Sarawak, Malaysia. The family members of the 2 index cases were subsequently identified and counseled regarding the disease and its genetic transmission. If they consented, they were tested for the enzyme and glycolipid levels. If the levels were found to be abnormal, the family members then underwent further genetic testing. Informed written consent was obtained from each participant in compliance with the requirements of good clinical practices. All participants were screened in Miri Hospital and Sarawak General Hospital between January 2018 and September 2020. The results of the tests were communicated to the participants and further counseling was given if needed.
In this study, we analyzed the data of our FD patients and aimed to elucidate their molecular, biochemical, and clinical characteristics. Data were collected in English using a standardized form (available online and on paper), quality checked, and reviewed. Demographic data collected included age at the time of FD diagnosis, sex, ethnicity, family pedigree, and consanguinity. Clinical data pertaining to FD comprised types of genetic mutation, α-gal A enzyme activity, lyso-GL3 biomarkers, renal profile (biochemistry, urinalysis, and imaging), cardiac parameters (12-lead electrocardiogram, pro-B-type natriuretic peptide [pro-BNP], and echocardiography) as well as findings from slit-lamp examination, audiogram, and sensory, neurological, and cutaneous examinations. Targeted neurological investigations were performed when clinical neurological manifestations were present. The types of treatment received, related adverse events, and clinical response were also included in the analysis.
α-gal A enzyme activity and concentration of lyso-GL3 biomarkers were tested using tandem mass spectrometry methods from dried blood spot samples. Disease-causing mutations in the
Transthoracic echocardiography studies were performed by the same experienced echocardiographer for all patients in the left lateral decubitus position, using Philips HD 15 and transducer S5-2 Adult sector. The HD15 included Philips QLAB quantification software, XRES image processing, and PureWare transducer crystal technology. The standard evaluation included M-mode, 2-dimensional (2-D), and Doppler studies according to the American Society of Echocardiography recommendations [4]. Left ventricle (LV) ejection fraction was calculated from apical 4-chamber views using Simpson’s method. The maximum LV wall thickness was defined as the greatest end-diastolic thickness in any myocardial segment. The LV end-diastolic diameter and end-systolic diameter were measured from M-mode and 2-D images obtained from parasternal long-axis views. The LV mass index was calculated using Devereux’s formula.
Case Reports
Index patient 1 was a 46-year-old man who presented with intermittent frothy urine and bilateral lower limb swelling when he was 35 years old in 2009. Investigations then showed proteinuria of 1 g/d; his blood pressure and renal function were normal. There was a progressive increase in proteinuria on follow-up, and a kidney biopsy was done 4 years later. The biopsy specimen was assessed using light microscopy and immunofluorescence study by a renal pathologist. Prominent podocytes with abundant foamy cytoplasm were found on histopatho-logical examination and staining for immunoglobulin (Ig) G, IgA, IgM, and complement were negative. Further clinical details are shown in Table 1.
Index patient 1 was found to have cornea verticillata (Figure 1). He did not take concomitant medication at any time in the medical history that might have induced such eye findings, such as chloroquine, amiodarone, and nonsteroidal anti-inflammatory drugs. Echocardiogram showed concentric left ventricular hypertrophy (LVH) with preserved ejection fraction. Family screening of this index patient was done and another 7 family members were found to have the enzyme abnormalities and genetic mutation (Figure 2). They were asymptomatic at the time of data collection.
Enzyme replacement therapy (ERT) was initiated in this patient in 2018, 5 years after the biopsy and after a delay of almost a decade since the first clinical manifestation of frothy urine and limb swelling. After 24 months of ERT, the agalsidase beta IgG antibody titer was 3200, while the neutralizing antibody was undetectable. The patient tolerated ERT well without developing hypersensitivity or adverse reaction. His proteinuria and renal function remained stable since the commencement of ERT.
Index patient 2 initially presented with frothy urine, oliguria, facial puffiness, and bilateral pedal edema. Investigations showed proteinuria of 8.45 g/d, and his blood pressure and serum glucose were normal. His renal function gradually deteriorated over 2 years and was subsequently diagnosed as end-stage renal disease of unknown cause. The patient also had intermittent nonspecific headache with 1 episode of generalized tonic-clonic seizure, with computed tomography of the brain showing multifocal infarcts in the background of small vessel ischemia and cerebral atrophy. Electroencephalogram was normal. A screening was done 7 years later when the patient presented to our hospital. He was found to have residual activity of α-gal A with normal lyso-Gb3 level and to carry a novel GLA mutation of c.548-5T>A. Renal biopsy was not done as his kidneys were shrunken, and he had undergone regular dialysis for years. The patient did not have any other manifestations apart from LVH on echocardiogram. Family screening identified another 5 family members who had the same genetic mutation (Figure 2). One of his brothers had chronic neuropathic pain involving bilateral lower extremities. However, he refused any further investigations due to the COVID-19 pandemic. Other family members were asymptomatic.
Discussion
The estimated incidence of FD is about 1 in 117 000 male live births [5]. However, newborn screening initiatives suggest that the incidence may be much higher, ranging from 1: 1500 in Taiwan to 1: 13 341 in Hungary [6,7]. Due to the fact that FD has a wide spectrum of heterogeneously progressive clinical phenotypes, it is likely that many patients, particularly females with FD are undiagnosed [8]. The disease spectrum ranges from the “classic” severe phenotype in males to an asymptomatic disease course observed in females, with a diversity in clinical manifestations between the extremes [9].
The diagnosis of classic FD is easily established in males (hemizygotes) because the enzyme activity is severely reduced or virtually absent in contrast with females (heterozygotes), in whom the enzyme activity in leukocytes may vary considerably in different cell types and tissues [8,9]. There is no clear association between clinical phenotypes and GLA enzyme activity among heterozygous females due to X chromosome inactivation, which may result in expression of α-gal A activity in the plasma or leukocytes within the normal range in up to 60% of women [10]. Thus, due to the overlap in leukocyte α-gal A activities between heterozygotes and noncarriers, mutation screening of GLA via gene sequencing should be part of the molecular genetic work-up in females, whereas enzyme activity measurement is likely adequate for establishing the diagnosis in males [10].
To date, no FD cases have been reported in Malaysia. This study summarized our very first clusters of FD by elucidating the molecular, biochemical, and clinical characteristics of Malaysian FD patients. We believe there are many more patients undiagnosed or unidentified in our nation due to a lack of awareness among clinicians, diversity in phenotypic spectrum, restricted accredited laboratory facilities, and limited resources. Hence, more needs to be done to improve the detection rate of this disorder in Malaysia.
The mechanism by which α-gal A deficiency and glycolipid accumulation result in such a wide variety of complications remains elusive. More than 900
For index patient 1, a variant within exon 4 of the
Another genetic mutation c.548-5T>A was discovered from our index patient 2. His α-gal A enzyme activity was slightly lower than the lower reference limit and his lyso-GL3 level was within the reference range. Pedigree analysis revealed nonclassical disease with similar biochemical findings as index patient 2 that may or may not have been caused by FD. To our knowledge, this
There are several debatable mutations associated with FD, for instance, p.D313Y and intronic variants, p.A143T and p.R112H variants. These have been described in literature as benign by some authors and as pathogenic by others [17,18]. Hence, we should depend on patients’ clinical findings, and not only on the genotype, when making a decision whether to commence ERT.
Studies reported that majority of FD patients develop antibodies toward agalsidase ERT, with high prevalence among male patients [19]. This might be attributed to most male FD patients having negligible residual enzyme activity, as was found in our index patient 1. Due to restricted access to laboratory testing and logistic difficulties, it is not feasible to monitor serial antibody titer and lyso-Gb3 levels. This is one of the limiting factors to study the relationship between ERT and the changes in lyso-Gb3 levels in correlation with the clinical outcome. It remains elusive whether long-term efficacy of ERT is hampered by these IgG antibodies and this warrants further studies to answer this hypothesis.
Clinicians should be aware that not all anti-drug antibodies (ADAs) possess neutralizing activity. It is of paramount importance to run neutralization assays to assess the effect of ADAs on clinical and biochemical outcomes [20]. Our index patient 1 showed IgG ADAs with a titer of 3000, but the neutralizing antibody was negative. This finding is consistent with the clinical response, highlighting the fact that ADA-positive status alone is not sufficient to justify the clinically relevant impact of ERT neutralization.
Cardiac involvement is one of the common causes of death in FD in both sexes [21,22], with concentric hypertrophy being the most frequently seen pattern [23]. It leads to progressive concentric, asymmetrical, or apical LVH, with heart failure and arrhythmia as the clinical sequelae. In patients with FD, LV mass and wall thickness increase with age [24]. Having hypertension should not preclude the clinical suspicion of FD, particularly in cases in which the hypertrophy distribution is disproportionate to the severity of hypertension [13,25]. In our study, we detected concentric LVH in 2 index patients who also demonstrated high levels of pro-BNP. Studies have shown that FD patients with elevated pro-BNP had higher risk of progression to cardiomyopathy and heart failure [26,27].
Proteinuria and reduced glomerular filtration rate are reported to be the early renal manifestations in FD patients with the classic phenotype, and they progressively result in end-stage renal disease in virtually all male patients [28].
It is of paramount importance to detect FD at an early stage and carry out thorough investigations to determine its possibly related comorbidities because it has been shown by recent evidence that the disease progression might be attenuated by early commencement of ERT [28]. For index patient 1, a delay in initiating ERT was due to the challenges posed in reaching the diagnosis and getting the access to treatment in a resource-limited setting. Sophisticated diagnostic assays are not readily available in our nation as local clinicians rarely screen for FD.
The true prevalence of FD in Malaysia is not known. Future systematic screening for the disease, particularly in high-risk population, is warranted, weighing the balance between the clinical benefits of early detection and the potential ethical and psychological ramifications of such a disorder.
Conclusions
This study outlines our local FD landscape and offers objective standards for forward planning. We hope this study may help identify any shortfalls in our health care system, discover areas of concern or potential research, predict future needs, and ensure cost-effective treatment for this group of patients. Shared responsibility among patients, families, care providers, public agencies, nongovernmental organizations, and insurance policy makers should be heightened so as to improve the care of our FD population. A local economic evaluation should be conducted to determine the best model of treatment for FD patients in Malaysia, taking the high diagnostic and treatment cost into consideration.
Figures
References:
1.. Laney DA, Bennett RL, Clarke V, Fabry disease practice guidelines: Recommendations of the national society of genetic counsellors: J Genet Counsel, 2013; 22; 555-64
2.. Brady RO, Gal AE, Bradley RM, Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency: N Engl J Med, 1967; 276(21); 1163-67
3.. Germain Dominique P, Fabry disease: Orphanet J Rare Dis, 2010; 5(1); 30
4.. Lang RM, Badano LP, Mor-Avi V, Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging: J Am Soc Echocardiogr, 2015; 28(1); 1-39 e14
5.. Meikle PJ, Hopwood JJ, Clague AE, Carey WF, Prevalence of lysosomal storage disorders: JAMA, 1999; 281(3); 249-54
6.. Hwu WL, Chien YH, Lee NC, Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A): Hum Mutat, 2009; 30; 1397-405
7.. Wittmann J, Karg E, Turi S, Newborn screening for lysosomal storage disorders in Hungary: JIMD Rep, 2012; 6; 117-25
8.. Eng CM, Fletcher J, Wilcox WR, Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry: J Inherit Metab Dis, 2007; 30(2); 184-92
9.. Wilcox WR, Oliveira JP, Hopkin RJ, Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry Registry: Mol Genet Metab, 2008; 93; 112-28
10.. Juchniewicz P, Kloska A, Tylki-Szymańska A, Female Fabry disease patients and X-chromosome inactivation: Gene, 2018; 641; 259-64
11.. Zarate YA, Hopkin RJ, Fabry’s disease: Lancet, 2008; 372; 1427-35
12.. Richards S, Aziz N, Bale S, Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology: Genet Med, 2015; 17(5); 405-24
13.. Dipple KM, McCabe ERB, Phenotypes of patients with “simple” Mendelian disorders are complex traits: Thresholds, modifiers, and system dynamics: Am J Hum Genet, 2000; 66; 1729-35
14.. Lukas J, Scalia S, Eichler S, Functional and clinical consequences of novel α-galactosidase A mutations in Fabry disease: Hum Mutat, 2016; 37(1); 43-51
15.. Greillier S, Daniel L, Caillaud C, First phenotypic description of a female patient with c.610 T>C variant of GLA: A renal-predominant presentation of Fabry disease: BMC Med Genet, 2020; 21; 137
16.. Van der Tol L, Smid BE, Poorthuis BJHM, A systematic review on screening for Fabry disease: Prevalence of individuals with genetic variants of unknown significance: J Med Genet, 2014; 51(1); 1-9
17.. du Moulin M, Koehn AF, Golsari A, The mutation p.D313Y is associated with organ manifestation in Fabry disease: Clin Genet, 2017; 92(5); 528-33
18.. Spada M, Pagliardini S, Yasuda M, High incidence of later-onset Fabry disease revealed by newborn screening: Am J Hum Genet, 2006; 79(1); 31-40
19.. Linthorst GE, Hollak CEM, Donker-Koopman WE, Strijland A, Aerts JMFG, Enzyme therapy for Fabry disease: Neutralizing antibodies toward agalsidase alpha and beta: Kidney Int, 2004; 66(4); 1589-95
20.. Lenders M, Schmitz B, Brand SM, Characterization of drug-neutralizing antibodies in patients with Fabry disease during infusion: J Allergy Clin Immunol, 2018; 141; 2289-92
21.. Waldek S, Patel MR, Banikazemi M, Life expectancy and cause of death in males and females with Fabry disease: Findings from the Fabry Registry: Genet Med, 2009; 11; 790-96
22.. Mehta A, Clarke JT, Giugliani R, Natural course of Fabry disease: Changing pattern of causes of death in FOS – Fabry Outcome Survey: J Med Genet, 2009; 46; 548-52
23.. Morrissey RP, Philip KJ, Schwarz ER, Cardiac abnormalities in Anderson-Fabry disease and Fabry’s cardiomyopathy: Cardiovasc J Afr, 2011; 22; 38-44
24.. Elliott P, McKenna WJ, Hypertrophic cardiomyopathy: Lancet, 2004; 363; 1881-91
25.. Sachdev B, Takenaka T, Teraguchi H, Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy: Circulation, 2002; 105; 1407-11
26.. Coats CJ, Parisi V, Ramos M, Role of serum N-terminal pro-brain natriuretic peptide measurement in diagnosis of cardiac involvement in patients with Anderson-Fabry disease: Am J Cardiol, 2013; 111; 111-17
27.. Seydelmann N, Liu D, Kramer J, High-sensitivity troponin: A clinical blood biomarker for staging cardiomyopathy in Fabry disease: J Am Heart Assoc, 2016; 5; e002839
28.. Kosch M, Koch HG, Oliveira JP, Enzyme replacement therapy administered during hemodialysis in patients with Fabry disease: Kidney Int, 2004; 66; 1279-82
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952428
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952854
Most Viewed Current Articles
07 Dec 2021 : Case report  22,760,204
22,760,204
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,117
176,117
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,604
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,593
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133