26 August 2021: Articles 
Polycythemia Vera Associated with Pulmonary Hypertension and Diffuse Large B-Cell Lymphoma: A Case Report
Challenging differential diagnosis, Rare disease, Rare coexistence of disease or pathology
Satoshi Kameda1ABCDEF, Fusako Sera1ABCDEF*, Kazuaki Sato2BCD, Masako Kurashige2BCD, Shuichiro Higo13E, Tomohito OhtaniDOI: 10.12659/AJCR.932956
Am J Case Rep 2021; 22:e932956






Abstract
BACKGROUND: Myeloproliferative neoplasms (MPNs), including polycythemia vera (PV), are associated with pulmonary hypertension (PH) and malignant lymphomas. Although the underlying mechanisms have not been completely clarified, it has been suggested that the Janus kinase 2 (JAK2) mutation, which is frequently identified in PV, can be involved in the development and/or progression of these distinct diseases in patients with MPNs. However, no reports have described the coexistence of PH and malignant lymphoma in patients with MPNs.
CASE REPORT: A 79-year-old man being treated for PV for 27 years and PH for 5 years was hospitalized due to severe dyspnea at rest. His soluble interleukin-2 receptor levels gradually increased and the chest computed tomography showed remarkable progression of the lung lesions and an enlargement of the mediastinal and axillary lymph nodes. A lymph node biopsy was performed and the patient was diagnosed with diffuse large B-cell lymphoma (DLBCL). Owing to his poor condition, chemotherapy was not initiated, and he died on the 89th day of hospitalization. The pathological autopsy revealed the destruction of alveolar structures with neoplastic space-occupying lesions of DLBCL. Multifactorial features of PH associated with MPNs, including the intimal thickening of pulmonary arteries accompanied by megakaryocytes and obstructed pulmonary arteries with organized thrombi in the lung tissue specimens, were observed. We found a JAK2 mutation based on a genetic analysis of the patient’s bone marrow.
CONCLUSIONS: We present the rare case of a patient who had PV with a JAK2 mutation, which coexisted with PH and DLBCL, and he developed severe refractory respiratory failure.
Keywords: Hypertension, Pulmonary, Janus Kinase 2, Lymphoma, Large B-Cell, Diffuse, myeloproliferative disorders, Polycythemia vera, Bone Marrow, Humans
Background
Myeloproliferative neoplasms (MPNs) are a group of hemato-logic disorders caused by the clonal proliferation of the bone marrow stem cells; these include polycythemia vera (PV), essential thrombocythemia, and primary myelofibrosis [1]. Although patients with MPNs have an increased risk of thrombohemorrhagic complications, these disorders are indolent in nature [2,3]. MPNs gradually worsen and eventually lead to bone marrow fibrosis or can transform to a blast crisis, including acute leukemia.
MPNs cause pulmonary hypertension (PH) through various mechanisms, and this is associated with a poor prognosis [4]. The current PH guidelines classify PH into 5 groups based on the underlying etiology and mechanism, and MPN-associated PH is classified as PH with unclear multifactorial mechanisms (Group 5) [5]. Most patients with MPNs, especially those with PV, have a mutation in the Janus kinase 2 (
Case Report
A 79-year-old man had an emergency admission to our hospital due to severe dyspnea at rest. He had a 27-year history of PV diagnosed at the age of 52 years, which had been well managed with hydroxycarbamide. In addition, he had a 5-year history of PH and a family history of gastric cancer. Right heart catheterization (RHC) performed at the time of diagnosis of the PH had revealed a mean pulmonary artery pressure (mPAP) of 39 mmHg, pulmonary artery wedge pressure of 9 mmHg, high cardiac output of 9.2 L/min, and a corresponding pulmonary vascular resistance (PVR) of 3.3 Wood units. He was diagnosed with PH associated with PV (Group 5) and was treated with a triple combination therapy of pulmonary vasodilators (bosentan, sildenafil, and beraprost). Anticoagulation with warfarin was initiated because a few localized ventilation-perfusion mismatches were noted in a ventilation/perfusion lung scan. A follow-up RHC performed 4 months before the present emergency admission revealed a persistent high cardiac output (9.3 L/min) with slight improvements in the hemodynamics (mPAP 32 mmHg and PVR 2.5 Wood units). However, he experienced a gradual worsening of the dyspnea on exertion.
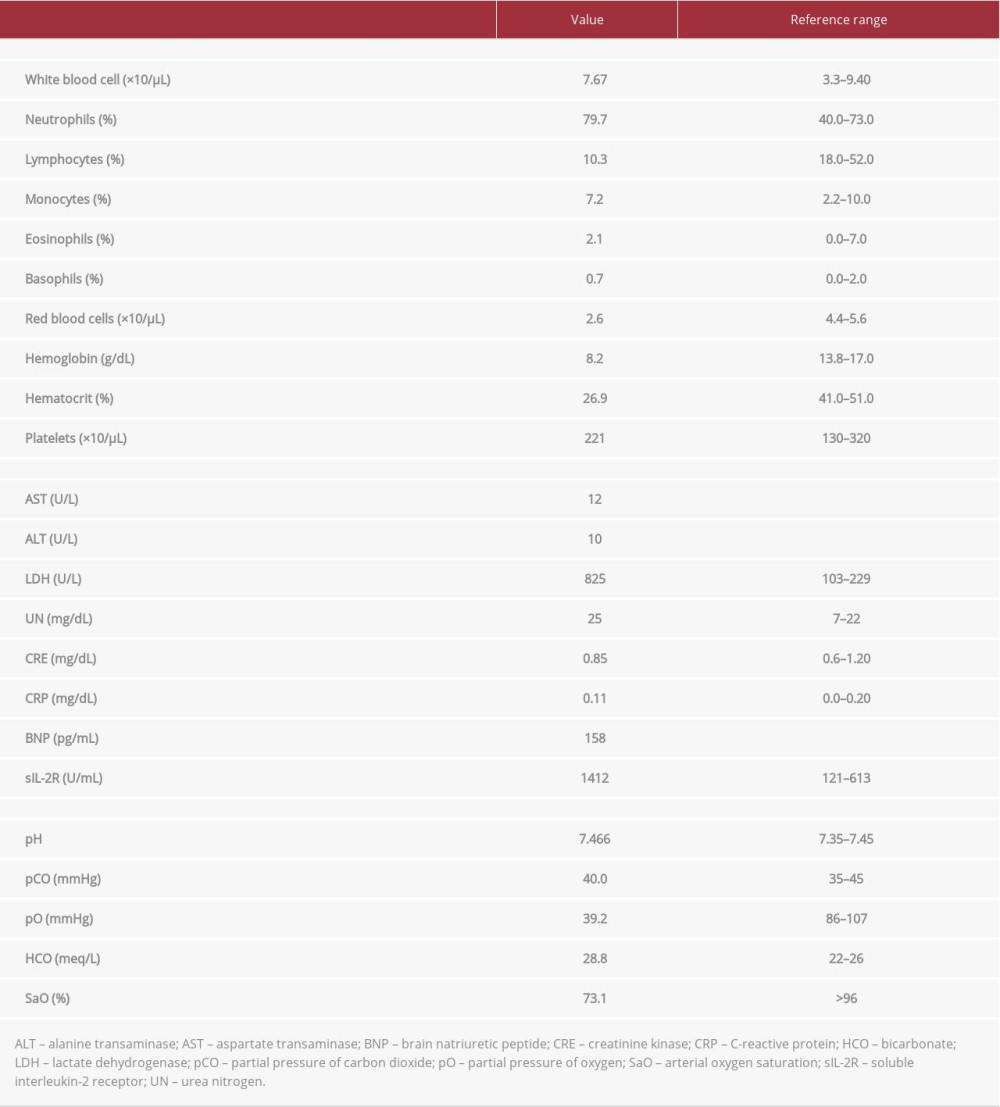
On admission, his blood pressure was 94/53 mmHg, pulse rate 98 beats/min, respiratory rate 36 breaths/min, oxygen saturation 73.1% while receiving 5 L/min of oxygen, and body temperature was 36.6°C. There was no palpable lymphadenopathy. The red blood cell count revealed anemia, although the white blood cells and platelet counts were within the normal ranges (Table 1). Blood biochemistry was almost normal, except for an elevated lactate dehydrogenase level, slightly elevated brain natriuretic peptide level, and mildly elevated soluble interleukin-2 receptor (sIL-2R) level. Chest radiography and computed tomography (CT) on admission revealed multiple nodules of variable sizes, and consolidations with ground-glass attenuation in both lung fields (Figure 1A, 1B). A bronchoscopy was planned to determine the cause of acute respiratory failure; however, we could not perform it, as the patient declined intubation. None of our therapeutic interventions (including antibiotics, steroids, and non-invasive positive pressure ventilation) were effective. His sIL-2R levels increased to 3239 IU/mL and a follow-up chest CT on the 70th day of hospitalization showed remarkable progression of the pulmonary lesions along with enlarged mediastinal and axillary lymph nodes (Figure 1C). An examination of the biopsy specimens obtained from the axillary lymph nodes revealed large lymphoid cells that were positive for CD20, CD79a, BCL-6, and MUM1 and negative for CD10, on immunohistochemical staining. He was diagnosed with diffuse large B-cell lymphoma (DLBCL).
Given his poor condition (performance status 4), chemotherapy was not initiated, and he died on the 89th day of hospitalization.
With the consent of his family, a pathological autopsy was performed, and this revealed the destruction of alveolar structures with neoplastic space-occupying lesions in both lungs (Figure 2A, 2B). Immunohistochemical staining of the specimen taken from the lung tumor confirmed the histological diagnosis of DLBCL, which was compatible with the findings from the lymph node biopsy specimen. A fibrous thickening of the intimal layer of the pulmonary arteries was seen (Figure 3A, 3B) in the lung tissue specimens. Plexiform lesions, which are advanced changes in the pulmonary artery and typically occur in cases of pulmonary arterial hypertension, were not detected. Some pulmonary arteries were obstructed by organized thrombi with many perforations, indicating that recanalization had commenced after pulmonary thrombosis (Figure 3C). Megakaryocytes were found in the lung tissue specimens, which was suggestive of extramedullary hematopoiesis (Figure 3D). Pulmonary veno-occlusive disease was not detected. Genetic analysis revealed that the bone marrow was positive for a
Committee of Osaka University Hospital. It conformed to the Ethical Guidelines of Medical and Health Research involving human subjects in Japan and all the principles outlined in the Declaration of Helsinki. We obtained informed consent from the patient’s family.
Discussion
We present the case of a patient with MPN-associated PH that developed into severe refractory respiratory failure. Although multifactorial PH was suspected to contribute to the patient’s chronic dyspnea, aggressive NHL was finally diagnosed as the cause of fatal respiratory failure. Although PH and NHL are entirely different disease entities, we speculate that the
In patients with MPNs, PH is an important complication that is associated with poor prognosis [4]. According to the current PH guidelines, PH associated with MPNs is classified as PH with unclear multifactorial mechanisms (Group 5) [5]. Various mechanisms for MPN-associated PH have been proposed, including thromboembolism, portopulmonary hypertension, high cardiac output, and the obstruction of pulmonary microvasculature by circulating megakaryocytes. Megakaryocytes that translocate from the bone marrow to the lungs can secrete vasoactive cytokines that could lead to the development of PH [10]. The pathological evaluation of our case showed intimal thickening in the pulmonary arteries accompanied by megakaryocyte infiltration and obstructed pulmonary arteries with organized thrombi, which were compatible with the previously reported multifactorial features of PH associated with MPNs. In addition,
In contrast, patients with MPNs have an increased risk of developing a second hematologic malignancy compared to the general population [9]. Although the subsequent hematologic neoplasms were mainly myeloid leukemia, the coexistence of MPNs with a lymphocytic proliferative neoplasm (LPN) has been reported [8,9,14]. The most common combination was MPNs and chronic lymphocytic leukemia; however, the coexistence of MPNs and NHL is rare [14]. A limited number of case reports describe the association between MPNs and NHL [8,15]. Popov et al reported a case of PV with a
Aberrant JAK-STAT signaling contributes to the pathogenesis of MPNs and can be involved in the development of lymphomas through the dysregulation of proliferation, differentiation, and apoptosis of hematopoietic cells [17]. The risk of developing LPN was significantly increased in patients with MPNs who had
Conclusions
We present a rare case of PV with coexisting PH and NHL. JAK inhibitors play an important role in the treatment of MPNs; however, their efficacy in the treatment of PH and NHL associated with MPNs has not been established. Therefore, further studies are required to elucidate the involvement of
Figures
References:
1.. Barbui T, Thiele J, Gisslinger H, The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion: Blood Cancer J, 2018; 8(2); 15
2.. Hultcrantz M, Bjorkholm M, Dickman PW, Risk for arterial and venous thrombosis in patients with myeloproliferative neoplasms: A population-based cohort study: Ann Intern Med, 2018; 168(5); 317-25
3.. Passamonti F, Rumi E, Pungolino E, Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia: Am J Med, 2004; 117(10); 755-61
4.. Dingli D, Utz JP, Krowka MJ, Unexplained pulmonary hypertension in chronic myeloproliferative disorders: Chest, 2001; 120(3); 801-8
5.. Galie N, Humbert M, Vachiery JL, 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT): Eur Respir J, 2015; 46(4); 903-75
6.. Levine RL, Wernig G, Role of JAK-STAT signaling in the pathogenesis of my-eloproliferative disorders: Hematology Am Soc Hematol Educ Program, 2006; 233–39; 510
7.. Tabarroki A, Lindner DJ, Visconte V, Ruxolitinib leads to improvement of pulmonary hypertension in patients with myelofibrosis: Leukemia, 2014; 28(7); 1486-93
8.. Vannucchi AM, Masala G, Antonioli E, Increased risk of lymphoid neoplasms in patients with Philadelphia chromosome-negative myeloproliferative neoplasms: Cancer Epidemiol Biomarkers Prev, 2009; 18(7); 2068-73
9.. Frederiksen H, Farkas DK, Christiansen CF, Chronic myeloproliferative neoplasms and subsequent cancer risk: A Danish population-based cohort study: Blood, 2011; 118(25); 6515-20
10.. Adir Y, Humbert M, Pulmonary hypertension in patients with chronic myeloproliferative disorders: Eur Respir J, 2010; 35(6); 1396-406
11.. Paulin R, Meloche J, Bonnet S, STAT3 signaling in pulmonary arterial hyper-tension: JAKSTAT, 2012; 1(4); 223-33
12.. Low AT, Howard L, Harrison C, Pulmonary arterial hypertension exacerbated by ruxolitinib: Haematologica, 2015; 100(6); e244-45
13.. Miyawaki H, Kioka H, Sato K, Long-term effects of the Janus kinase 1/2 inhibitor ruxolitinib on pulmonary hypertension and the cardiac function in a patient with myelofibrosis: Intern Med, 2020; 59(2); 229-33
14.. Hauck G, Jonigk D, Kreipe H, Simultaneous and sequential concurrent myeloproliferative and lymphoproliferative neoplasms: Acta Haematol, 2013; 129(3); 187-96
15.. Rumi E, Passamonti F, Elena C, Increased risk of lymphoid neoplasm in patients with myeloproliferative neoplasm: A study of 1,915 patients: Haematologica, 2011; 96(3); 454-58
16.. Popov VM, Dobrea CM, Popescu M, Diffuse large B-cell lymphoma and polycythemia vera discovered at the onset – a rare association and its possible importance in lymphoma prognosis: Rom J Morphol Embryol, 2016; 57(1); 313-18
17.. Rumi E, Barate C, Benevolo G, Myeloproliferative and lymphoproliferative disorders: State of the art: Hematol Oncol, 2020; 38(2); 121-28
18.. Marchetti M, Carobbio A, Capitoni E, Lymphoproliferative disorders in patients with chronic myeloproliferative neoplasms: A systematic review: Am J Hematol, 2018; 93(5); 698-703
Figures
In Press
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953068
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133