20 October 2021: Articles 
A 46-Year-Old Woman with Primary Infertility and a Diagnosis of Microcystic Stromal Tumor of the Ovary Confirmed by Histology and Gene Sequencing: A Case Report and Review of the Literature
Challenging differential diagnosis, Rare disease, Educational Purpose (only if useful for a systematic review or synthesis)
Maria A. Arafah1ABCDEF*, Layla AlBreacanDOI: 10.12659/AJCR.933528
Am J Case Rep 2021; 22:e933528






Abstract
BACKGROUND: Microcystic stromal tumor (MCST) of the ovary is a rare entity with distinct pathological and molecular features. However, a lack of awareness of ovarian MCST can lead to delayed diagnosis or misdiagnosis. We present a case of ovarian MCST and review all previously reported cases and discuss their clinical and pathological characteristics.
CASE REPORT: A 46-year-old woman with primary infertility due to polycystic ovary syndrome presented with bleeding and pain. Radiological images showed a complex solid and cystic adnexal mass. Microscopically, the tumor was lobulated with cellular regions separated by fibrous plaques and small anastomosing cysts, consistent with an ovarian MCST. The tumor cells showed positive staining for vimentin, CAM 5.2, CD10, β-Catenin, CD99, and cyclin D1. Genetic sequencing showed a point mutation in the CTNNB1 gene, with no mutations in the APC, BRCA1, and BRCA2 genes. The patient underwent surgery and was disease-free at 24 months after her initial diagnosis.
CONCLUSIONS: The diagnosis of ovarian MCST should consider the differential diagnosis of cystic tumors of the ovary. Further research is encouraged to elucidate the various molecular pathways involved in the pathogenesis of this tumor and to determine its optimal treatment and long-term prognosis.
Keywords: Ovarian Neoplasms, CTNNB1 Protein, Human, Genes, APC, Female, Humans, Infertility, Sex Cord-Gonadal Stromal Tumors, Vimentin
Background
Ovarian microcystic stromal tumor (MCST) was first reported in 2009 by Irving and Young [1] and was then included as a pure ovarian stromal tumor in the 2014 World Health Organization Classification of Tumors of the Female Reproductive Organs [2]. Ovarian MCSTs have distinctive morphological features, including microcysts and cellular lobules with an intervening fibrous, or sometimes hyalinized stroma. They lack the morphological features of other sex cord-stromal tumors and do not show any germ cell, teratomatous, or epithelial elements. They are also characterized by unique immunohistochemical and molecular profiles [1,3].
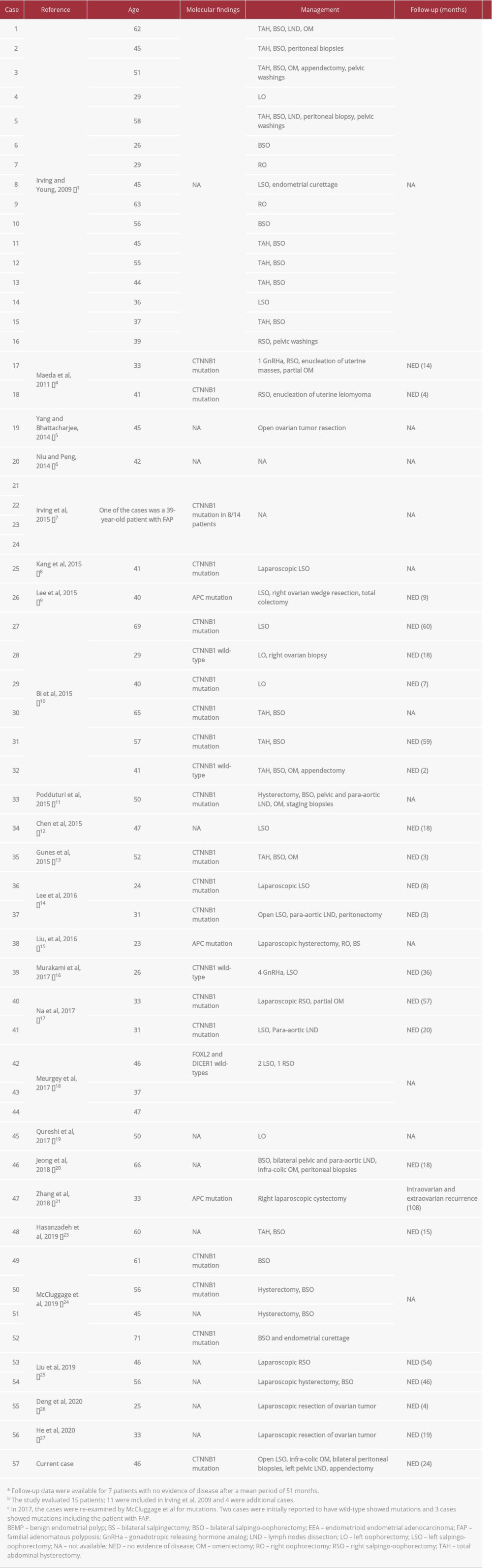
Fewer than 60 cases of ovarian MCSTs have been reported to date [1,4–27] all of which were in adults, had a mean size of <10 cm, and had benign outcomes, except for a case of pelvic recurrence [21]. We present a case of an ovarian MCST in a 46-year-old woman with primary infertility due to polycystic ovary syndrome. We also discuss the histopathological, radiological, immunophenotypical, and molecular features of this tumor and review all previously reported cases.
Case Report
A 46-year-old woman presented with intermittent vaginal bleeding and moderate abdominal pain for 3 days. She had a history of primary infertility and polycystic ovaries, for which she received various fertility regimens and underwent a series of ultrasound imaging for 3 years. She also underwent a myomectomy for a uterine fibroid 6 years before her presentation. She had no family history of malignant tumors. The patient had a body mass index of 36.85 kg/m2 and a slightly tender abdomen with no palpable inguinal lymph nodes on physical examination. Her vital signs were stable upon admission, and all laboratory investigations were within normal limits, including a complete blood count, coagulation profile, urea and electrolyte panel, renal and liver function tests, and tumor markers (CA 125, CA 15-3, CA 19-9, CEA, AFP, and β-HCG).
Pelvic ultrasound showed a large complex left adnexal mass, measuring 15.3×12.3×10.6 cm, with cystic and solid components. Two small intramural fibroids were also noted. Magnetic resonance imaging (MRI) of the pelvis showed a complex multiloculated cystic mass with few enhancing solid areas, measuring 17×15×9 cm (Figure1A, 1B). The uterus was displaced with multiple intramural fibroids and showed diffuse adenomyosis. A large area of loculated fluid was also seen in the pouch of Douglas, measuring 7×6.5×7.5 cm, displacing the rectum posteriorly. Radiologically, these findings were likely to represent a malignant epithelial ovarian neoplasm. The patient was scheduled for surgery with an intraoperative consultation, which suggested at the time a sex cord-stromal tumor. The patient underwent an open left salpingo-oophorectomy, infra-colic omentectomy, bilateral peritoneal biopsies, left pelvic lymph nodes dissection, and an appendectomy. Fluid from the pouch of Douglas was taken for cytological examination and was negative for malignant cells.
Macroscopically, the ovarian mass weighed 935 g and measured 17.5×16×6.4 cm. It was firm with a smooth outer surface. Serial slicing showed a complex partially solid and partially cystic mass. The cystic areas were filled with a clear fluid, while the solid areas were pale tan and homogenous. No papillary projections were seen.
Microscopically, the tumor was lobulated with cellular regions separated by fibrous plaques (Figure 2A) and small anastomosing cysts (Figure 2B). The neoplastic cells were round with pale finely granular eosinophilic cytoplasm and round to oval nuclei (Figure 2C, 2D). Small nucleoli were occasionally noted, and mitoses were absent. The other specimens were histologically unremarkable.
The tumor cells showed positive staining for vimentin, CAM 5.2, CD10, β-Catenin, CD99 (paranuclear dot-like pattern), and cyclin D1 (Figure 3A–3F). Around 5% to 10% of cells showed weak nuclear positivity for progesterone receptors. All other markers were negative, including inhibin, calretinin, PLAP, EMA, cytokeratin 20, cytokeratin 7, melan A, chromogranin, synaptophysin, AFP, WT-1, and estrogen receptors. The microscopic findings and immunohistochemical results were in keeping with an MCST of the ovary. Genetic sequencing showed a point mutation in the
The patient was discharged in good health on the third day after surgery, and she was disease-free 24 months after her initial diagnosis. She is still following up with her gynecologist for primary infertility and general surgery for a future sleeve gastrectomy. A written informed consent for patient information and images to be published was obtained from the patient.
Discussion
The first detailed description of an ovarian MCST was published in 2009 by Irving and Young, based on 16 uncategorized tumors of the ovary [1]. In 2011, Maeda et al reported β-catenin nuclear expression and
A total of 56 cases of ovarian MCST have been reported to date (Table 1). All cases were in adults, with a mean age of 44 years (range: 23–71 years) [1,4–27]. All cases were unilateral, with more on the left side, had a mean size of <10 cm, and lacked hormonal manifestations [1,4–27]. More than half of the patients presented with a mass (28/52, 53.85%), followed by pelviabdominal pain, discomfort, or fullness (14/52, 26.92%), or their tumors were discovered incidentally during routine checkups or while other unrelated symptoms were being investigated (10/52, 19.23%). More than half of the patients (24/51, 47%) had associated gynecological (12/24, 50%) or non-gynecological (12/24, 50%) disorders. The clinical features of familial adenomatous polyposis were present in 4 patients [9,15,21,22], and 5 patients had a history of malignancy (4 endometrioid endometrial adenocarcinomas, 1 contralateral ovarian endometrioid adenocarcinoma, and 1 colonic adenocarcinoma) [1,6,24]. Tumor markers were evaluated in 19 cases with limited data and undetermined clinical significance; however, an elevation in CA 125 was reported in 4 patients [5,11,14,17].
Ovarian MCSTs are well-circumscribed lesions, and they are usually solid and cystic (29/50, 58%) on macroscopic and/or radiologic examinations. Sonographically, ovarian MCST appears as a hypovascular complex or anechoic cystic mass [14,20]. It is seen as a heterogeneous mass with hyperattenuating and hypoattenuating components on computed tomography scans. Positron emission tomography shows FDG uptake within the solid areas [20]. On MRI, the cystic areas show high-signal intensity on T2-weighted MRI images and iso-signal intensity on T1-weighted images [16].
Microscopically, MCST is a cellular lobulated neoplasm with intervening fibrous, sometimes hyalinized, plaques or bands and, as the name indicates, anastomosing microcysts. The cells are usually monotonous with round to oval nuclei, small nucleoli, and a finely granular pale eosinophilic cytoplasm. Occasionally, the neoplastic cells are spindle-shaped or show a diffuse corded and nested growth pattern either focally, predominantly, or exclusively [24]. Multinucleated cells and cells with bizarre pleomorphic degenerative (symplastic-like) nuclei can infrequently occur, and the latter can account for up to 50% of the tumor [10,12,18,27]. Mitoses are rare in most cases (0–2 in 10 high-power fields), with no reported atypical forms [10,18]. The stromal nature of ovarian MCST was supported by ultrastructural findings, including the lake of Reinke crystals, true lumina, cilia, microvilli, desmosomes, and cellular junctions [5].
The use of immunohistochemical staining helps differentiate ovarian MCST from its mimickers. The neoplastic cells in MCST show an aberrant nuclear expression for β-catenin and cyclin D1 [1,4–27]. They are usually positive for vimentin, CD10, WT-1, FOXL2, and SF-1 [1,4–27]. Similar to our case, Bi et al reported paranuclear dot-like staining for CD99 in 3 of the 5 cases examined in their series and progesterone-receptor expression in around 5% of cells in 2 cases [10]. CD56 was reported as positive in 10% of tumor cells in 1 case of ovarian MSCT [10]. However, a more diffuse, stronger progesterone-receptor and CD56 staining was displayed in another study [12]. Deng et al reported patchy positive staining for synaptophysin and diffuse positive staining for androgen receptors in 2 cases [24,26]. Reports showed that ovarian MCST is negative for inhibin, calretinin, EMA, E-cadherin, CD117, PLAP, SALL-4, desmin, and melan A [1,4-27].
The differential diagnosis encompasses a wide range of ovarian tumors. Thecomas usually occur in older women and present with estrogenic effects. They do not show the microcystic pattern of ovarian MCSTs and are positive for calretinin and inhibin [1,11]. Although an ovarian solid pseudopapillary neoplasm (SPN) shows an aberrant β-catenin nuclear expression and
On a molecular level, essentially all ovarian MCSTs are thought to have heterozygous point mutations in the
Ovarian MCST was treated surgically in a total of 36 patients (66.67%) undergoing oophorectomy (with or without salpingectomy) or cystectomy and 18 patients undergoing hysterectomy (including bilateral or unilateral salpingo-oophorectomy) with or without omentectomy, lymph node dissection, or appendectomy (33.33%) (Table 1). Two patients received gonadotropin-releasing hormone analog injections but showed tumor size progression and eventually underwent unilateral salpingo-oophorectomy [4,16].
Lymph node dissection is not mandatory in cases of sex cord-stromal tumors and germ cell tumors. The lymph nodes in our patient were not palpable bilaterally, and they were not suspicious radiologically; however, since the diagnosis was not confirmed during the surgery, an ipsilateral lymph node dissection was performed. If lymph nodes are involved with a sex cord-stromal tumor or germ cell tumor, chemotherapy would be indicated, regardless of the status of the contralateral lymph nodes. An appendectomy, although it may not have been necessary, was performed in our patient because an epithelial mucinous tumor was still among the radiological differential diagnoses.
Regardless of the chosen surgical management, all reported cases of ovarian MCST showed a good prognosis and uneventful outcomes (mean follow-up of 26.45 months), except for 1 patient who presented with a recurrence 9 years after her initial diagnosis [21]. The literature is limited regarding the optimal follow-up approach for these patients; however, we recommend annual pelvic ultrasound screening, followed by MRI if indicated.
Conclusions
We described a case of ovarian MCST and outlined the clinical, radiological, and pathological features of this tumor in view of all the reported cases in the literature. This tumor should be considered when there is presentation of cystic ovarian tumors. Furthermore, the present data provide insight into this tumor and encourage further research into its pathogenesis, behavior, treatment, and long-term prognosis.
Figures
References:
1.. Irving JA, Young RH, Microcystic stromal tumor of the ovary: Report of 16 cases of a hitherto uncharacterized distinctive ovarian neoplasm: Am J Surg Pathol, 2009; 33(3); 367-75
2.. Kurman R, Carcangiu M, Herrington C, Young R: World Health Organization Classification of Tumours of Female Reproductive Organs, 2014, Geneva, International Agency for Research on Cancer
3.. : Female Genital Tumours, 2020, Geneva, International Agency for Research on Cancer
4.. Maeda D, Shibahara J, Sakuma T, β-catenin (CTNNB1) S33C mutation in ovarian microcystic stromal tumors: Am J Surg Pathol, 2011; 35(10); 1429-40
5.. Yang M, Bhattacharjee MB, Ovarian microcystic stromal tumor: Report of a new entity with immunohistochemical and ultrastructural studies: Ultrastruct Pathol, 2014; 38(4); 261-67
6.. Niu S, Peng Y, Distinct immunophenotypic features of ovarian microcystic stromal tumor: Am J Clin Pathol, 2014; 142; A239
7.. Irving JA, Lee C-H, Yip S, Microcystic stromal tumor: A distinctive ovarian sex cord-stromal neoplasm characterized by FOXL2, SF-1, WT-1, Cyclin D1, and β-catenin nuclear expression and CTNNB1 mutations: Am J Surg Pathol, 2015; 39(10); 1420-26
8.. Kang YN, Cho CH, Kwon SY, Microcystic stromal tumor of the ovary with mutation in exon 3 of β-catenin: A case report: Int J Gynecol Pathol, 2015; 34(2); 121-25
9.. Lee SH, Koh YW, Roh HJ, Ovarian microcystic stromal tumor: A novel extracolonic tumor in familial adenomatous polyposis: Genes Chromosomes Cancer, 2015; 54(6); 353-60
10.. Bi R, Bai Q-M, Yang F, Microcystic stromal tumour of the ovary: Frequent mutations of β-catenin (CTNNB1) in six cases: Histopathology, 2015; 67(6); 872-79
11.. Podduturi V, Tran T, Champion KJ, Microcystic stromal tumor of the ovary: A case report of a newly described ovarian neoplasm with a β-catenin (CTNNB1) G34E mutation: Int J Gynecol Pathol, 2015; 34(6); 541-45
12.. Chen Q, Lu W, Lv W, Overlap of microcystic stromal tumor and primary solid pseudopapillary neoplasm of the ovary: Int J Clin Exp Pathol, 2015; 8(9); 11792-97
13.. Gunes P, Kir G, Yilmaz İ, Küçükodaci Z, Coexistence of microcystic stromal tumor of the ovary with mutation of β-catenin and contralateral mucinous cystadenoma: Int J Gynecol Pathol, 2015; 34(6); 546-50
14.. Lee JH, Kim H-S, Cho NH, Genetic analysis of ovarian microcystic stromal tumor: Obstet Gynecol Sci, 2016; 59(2); 157-62
15.. Liu C, Gallagher RL, Price GR, Ovarian microcystic stromal tumor: A rare clinical manifestation of familial adenomatous polyposis: Int J Gynecol Pathol, 2016; 35(6); 561-65
16.. Murakami M, Wroblewski J, Kawagoe H, Microcystic stromal tumor resected by laparoscopic surgery: Gynecol Minim Invasive Ther, 2017; 6(3); 135-38
17.. Na K, Kim EK, Jang W, Kim H-S, CTNNB1 mutations in ovarian microcystic stromal tumors: Identification of a novel deletion mutation and the use of pyrosequencing to identify reported point mutation: Anticancer Res, 2017; 37(6); 3249-58
18.. Meurgey A, Descotes F, Mery-Lamarche E, Devouassoux-Shisheboran M, Lack of mutation of DICER1 and FOXL2 genes in microcystic stromal tumor of the ovary: Virchows Arch, 2017; 470(2); 225-29
19.. Qureshi A, Hassan M, Mamoon N, Sex cord stromal tumours of the ovary, experience at Shifa International Hospital Islamabad: J Pak Med Assoc, 2017; 67(7); 1107-8
20.. Jeong D, Hakam A, Abuel-Haija M, Chon HS, Ovarian microcystic stromal tumor: Radiologic-pathologic correlation: Gynecol Oncol Rep, 2018; 25; 11-14
21.. Zhang Y, Tao L, Yin C, Ovarian microcystic stromal tumor with undetermined potential: Case study with molecular analysis and literature review: Hum Pathol, 2018; 78; 171-76
22.. McCluggage WG, Irving JA, Chong A-S, Ovarian microcystic stromal tumors are characterized by alterations in the beta-catenin-APC pathway and may be an extracolonic manifestation of familial adenomatous polyposis: Am J Surg Pathol, 2018; 42(1); 137-39
23.. Hasanzadeh M, Bazmi F, Malakuti P, An ovarian mass with microcystic stromal tumor: A rare case report: J Obstet Gynecol Cancer Res, 2019; 4(3); 127-30
24.. McCluggage WG, Chong A-S, Attygalle AD, Expanding the morphological spectrum of ovarian microcystic stromal tumour: Histopathology, 2019; 74(3); 443-51
25.. Liu J, Hou Y, Bao L, Ovarian microcystic stromal tumors: Clinical, radiological, and pathological studies of two cases: Int J Clin Exp Pathol, 2019; 12(6); 2241-48
26.. Deng L, Feng D, Liang J, Ovarian microcystic stromal tumor: A case report and literature review: Front Med (Lausanne), 2020; 7; 58
27.. He Y, Xu L, Feng M, Wang W, Ovarian microcystic stromal tumor with significant bizarre nuclei: A case report: Medicine (Baltimore), 2020; 99(34); e21841
28.. Cheuk W, Beavon I, Chui DTY, Chan JKC, Extrapancreatic solid pseudopapillary neoplasm: Report of a case of primary ovarian origin and review of the literature: Int J Gynecol Pathol, 2011; 30(6); 539-43
29.. Deshpande V, Oliva E, Young RH, Solid pseudopapillary neoplasm of the ovary: A report of 3 primary ovarian tumors resembling those of the pancreas: Am J Surg Pathol, 2010; 34(10); 1514-20
30.. Rabban JT, Karnezis AN, Devine WP, Practical roles for molecular diagnostic testing in ovarian adult granulosa cell tumour, Sertoli-Leydig cell tumour, microcystic stromal tumour and their mimics: Histopathology, 2020; 76(1); 11-24
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952107
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952658
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953243
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952989
Most Viewed Current Articles
07 Dec 2021 : Case report
22,697,854
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,914
174,914
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,026
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,962
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133