26 January 2022: Articles 
Using a Minimal Parameter Set for Early Diagnosis of Hemophagocytic Lymphohistiocytosis in Non-European Children
Challenging differential diagnosis
Yu Nakashima1BDEF, Hiroshi Koga1AEF*DOI: 10.12659/AJCR.934037
Am J Case Rep 2022; 23:e934037






Abstract
BACKGROUND: Children with hemophagocytic lymphohistiocytosis require rapid diagnosis for timely treatment. However, diagnostic delays may arise in settings with limited clinical resources. To address this issue, a simplified rule for diagnosing hemophagocytic lymphohistiocytosis has recently been proposed. We retrospectively applied this diagnostic rule in 2 infants to evaluate its generalizability to non-European children.
CASE REPORT: We present 2 cases of hemophagocytic lymphohistiocytosis, involving an Asian neonate with secondary hemophagocytic lymphohistiocytosis subsequent to echovirus infection and an African infant with familial hemophagocytic lymphohistiocytosis caused by PRF1 mutation. Limitations on time and clinical resources prevented tissue biopsy and measurement of natural killer cell activity in either case at our center. The Asian case did not meet HLH-2004 criteria, but both cases met a rapid diagnostic rule on admission to our center. Both cases were transported to a tertiary center and diagnosed with hemophagocytic lymphohistiocytosis based on HLH-2004 criteria. Although treatment suppressed disease activity, the Asian neonate died of multiple-organ failure at age 6 months. The African infant remains in remission after allogenic cord blood stem cell transplantation.
CONCLUSIONS: A simplified diagnostic rule for hemophagocytic lymphohistiocytosis may be useful for early diagnosis of hemophagocytic lymphohistiocytosis in Asian and African children, especially in resource-limited clinical settings. Further investigation is required to elucidate whether early diagnosis with a simplified diagnostic rule improves treatment outcomes for children with hemophagocytic lymphohistiocytosis.
Keywords: Clinical Decision Rules, Infant, Lymphohistiocytosis, Hemophagocytic, Early Diagnosis, Humans, Mutation
Background
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disease that requires rapid diagnosis and treatment. However, the HLH-2004 criteria include tissue biopsy and diagnostic assay for natural killer (NK) cell activity, both of which require time-consuming tests [1] that may be difficult to perform in a timely manner in clinical practice. In fact, a recent large case series study demonstrated that tissue biopsy was examined in 77 of 101 Egyptian children (76%) with HLH, while NK-cell activity was examined in 0 (0%) [2]. This diagnostic dilemma may be common around the world, particularly in facilities with limited clinical resources. To address this issue, a simplified and rapid prediction rule for the diagnosis of HLH has been proposed [1].
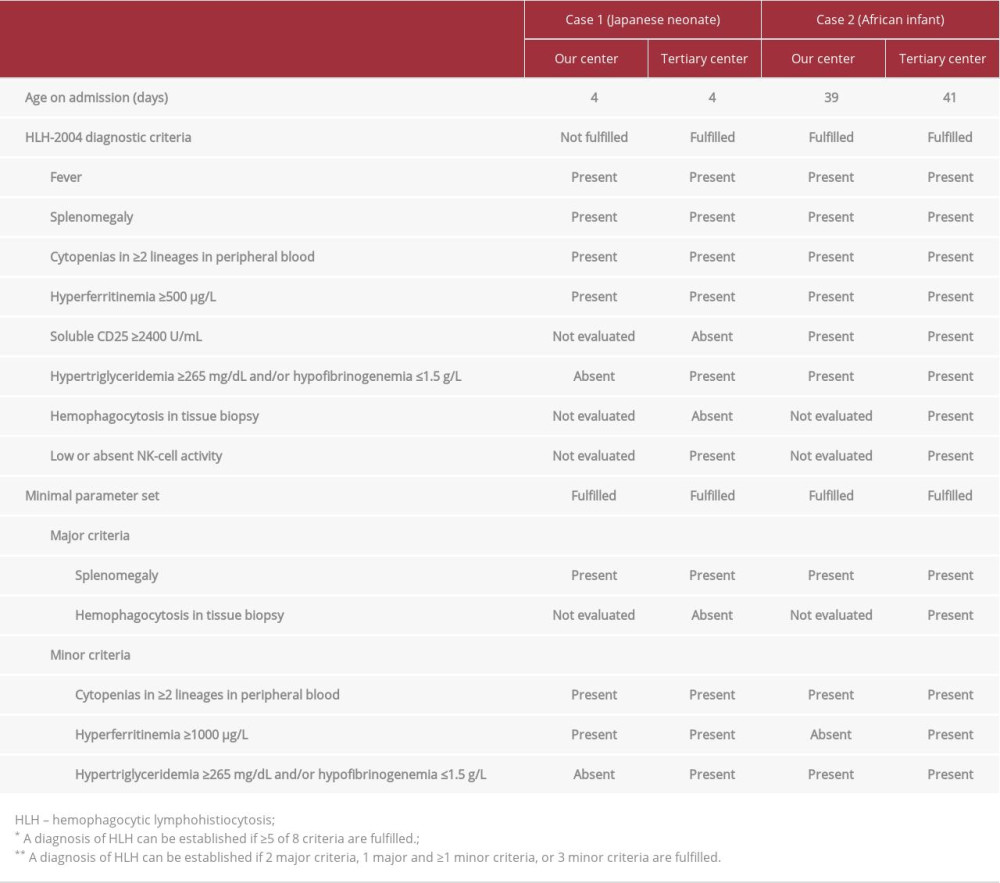
We recently encountered 2 cases of HLH, involving an Asian child and an African child. We used the clinical data from these cases to retrospectively evaluate the diagnostic utility of the HLH-2004 criteria [3] in comparison with a recently proposed rapid diagnostic rule of HLH, as a minimal parameter set [1], between 2 time points: on admission to our non-specialized center; and on transportation to a specialized tertiary center (Table 1). Through this report of 2 cases, we evaluated the clinical utility of a minimal parameter set for early diagnosis of HLH in non-European populations.
Case Reports
CASE 1: AN ASIAN NEONATE:
The first case involved a male Japanese neonate admitted to our center at age 4 days with fever (39.5°C) and splenomegaly. Initial blood testing showed: absolute neutrophil count, 0.99×109/L; platelet count, 98×109/L; ferritin, 21 400 μg/L; triglycerides, 65 mg/dL; and fibrinogen, 1.8 g/L. Assays for soluble CD25 or NK-cell activity and tissue biopsy were not performed. Clinical findings did not meet the HLH-2004 criteria, but the patient was transferred to a tertiary center on suspicion of HLH. Hypofibrinogenemia and low NK-cell activity were confirmed after transportation, and echoviruses 6 and 11 were isolated from cerebrospinal fluid, pharyngeal swab, and stool. No known mutations associated with HLH were identified, so secondary HLH was diagnosed. Treatment was initiated with corticosteroid, cyclosporine A, and intravenous immunoglobulin, which suppressed disease activity. However, the patient died of multiple-organ failure at age 6 months while awaiting liver transplantation for hepatic failure.
CASE 2: AN AFRICAN INFANT:
The second case involved a male Nigerian infant admitted to our center at age 39 days with fever (38.8°C) and splenomegaly.
His sister had died of unknown causes at age 1 month. Initial blood testing showed: absolute neutrophil count, 0.88×109/L; platelet count, 97×109/L; ferritin, 572 μg/L; triglycerides, 68 mg/dL; fibrinogen, 1.4 g/L; and soluble CD25, 9153 U/mL. He was transferred to a tertiary center under a presumptive diagnosis of HLH. After transportation, homozygous c.50delT mutations in the PRF1 gene were confirmed [3] and familial HLH type 2 was diagnosed. Allogenic cord blood stem cell transplantation was performed and the patient remains in remission to date, at age 4 years.
DIAGNOSTIC VALIDITY OF A MINIMAL PARAMETER SET:
We retrospectively applied a minimal parameter set to each case to evaluate the diagnostic validity at the 2 time points of admission to our non-specialized center and admission to a specialized tertiary center (Table 1). Case 1 did not meet the HLH-2004 criteria on admission to our center, but both cases met the minimal parameter set.
Discussion
To facilitate early diagnosis and treatment of HLH, simplified and rapid diagnostic criteria have been proposed [1]. A minimal parameter set was developed based on datasets from 5 medical centers in The Netherlands and validated by a dataset from Johns Hopkins Hospital. Because the HLH-related genetic background and clinical manifestations are known to differ among populations [4,5], the generalizability of that minimal parameter set to non-European populations has not previously been confirmed. We therefore evaluated the utility of the minimal parameter set for early diagnosis in African and Asian children. Although this evaluation included only 2 cases, the minimal parameter set seemed to offer external validity for these non-European cases.
In an epidemiological study of pediatric HLH by Elsharkawy et al, the 3-year overall survival rate was 30% among 101 Egyptian children with HLH (22% with primary HLH; 61% with secondary HLH) [2]. This observation emphasizes the need to improve the survival of children with HLH. Early diagnosis of HLH can lead to timely decision-making regarding treatment, which may be associated with improved survival. Children with poor general condition, bicytopenia, and/or splenomegaly require careful evaluation for HLH.
Some limitations to our study merit consideration. First, this was a retrospective evaluation of a diagnostic rule with an extremely limited sample size. Second, the diagnostic utility was evaluated in a clinical resource-limited setting. To demonstrate the enhanced generalizability of the minimal parameter set and any effects on clinical outcomes, further large-scale studies are warranted.
Conclusions
The rapid diagnostic rule seems beneficial for early diagnosis of Asian and African children with HLH. Further study is required to confirm whether early diagnosis and treatment with the rapid diagnostic rule improve treatment outcomes for children with HLH.
References:
1.. Smits BM, van Montfrans J, Merrill SA, A minimal parameter set facilitating early decision-making in the diagnosis of hemophagocytic lymphohistiocytosis: J Clin Immunol, 2021; 41; 1219-28
2.. Elsharkawy A, Assem H, Salama M, Clinical characteristics and outcomes of 101 children with hemophagocytic lymphohistiocytosis: A four-year single-center experience from Egypt: Pediatr Hematol Oncol, 2021; 38; 194-207
3.. Henter JI, Horne A, Arico M, Hlh-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis: Pediatr Blood Cancer, 2007; 48; 124-31
4.. Jordan MB, Allen CE, Weitzman S, How I treat hemophagocytic lymphohistiocytosis: Blood, 2011; 118; 4041-52
5.. Chinn IK, Eckstein OS, Peckham-Gregory EC, Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis: Blood, 2018; 132; 89-100
In Press
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953068
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133