09 November 2021: Articles 
Prompt Recognition of Hemophagocytic Lymphohistiocytosis in an Afebrile Patient with Lupus and Bacteremia
Challenging differential diagnosis, Management of emergency care, Rare coexistence of disease or pathology
Joshua Hardage1ADEF, Natalie B. Otto1ADEF, Joanne SkaggsDOI: 10.12659/AJCR.934092
Am J Case Rep 2021; 22:e934092






Abstract
BACKGROUND: Hemophagocytic lymphohistiocytosis (HLH) is a rare clinical syndrome characterized by dysregulated immune system activation and hyperinflammation. Primary HLH is inherited and almost exclusively seen in childhood, while secondary HLH is mainly seen in adults and has a wide variety of triggering factors, including infection, malignancy, autoimmune disease, and immunosuppression. Due to nonspecific presentation, the differential diagnosis for HLH is equally wide. We present a case of secondary HLH involving undiagnosed systemic lupus erythematosus and bacteremia.
CASE REPORT: A 43-year-old man with a history of discoid lupus presented with 1 month of weakness, epistaxis, shortness of breath, anorexia, and weight loss. He took no medications and did not follow with a primary care physician. Workup revealed leukopenia and thrombocytopenia, severely elevated ferritin, severe acute kidney injury, class II lupus nephritis on renal biopsy, hemophagocytic histiocytes on bone marrow biopsy, and other findings of end-organ damage. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Diagnosis of HLH occurred on the third day of admission. Our patient improved rapidly on high-dose corticosteroids, hydroxychloroquine, anakinra, tocilizumab, and low-dose etoposide as well as concomitant antibiotic therapy.
CONCLUSIONS: Despite having a diagnosis of discoid lupus, our patient was not established with a primary care physician and did not take any medications. This resulted in unknown smoldering systemic lupus erythematosus, which, possibly in conjunction with bacteremia, triggered a nearly fatal disease. We discuss the importance of primary care in disease management, the differentiation of sHLH from other diagnoses, HLH treatment, and the laboratory evaluation of sHLH.
Keywords: Acute Kidney Injury, Internal Medicine, Lupus Erythematosus, Cutaneous, Lupus Erythematosus, Systemic, Lymphohistiocytosis, Hemophagocytic, Primary Health Care, Bacteremia, Humans, Lupus Erythematosus, Discoid, Male, Staphylococcus aureus
Background
Hemophagocytic lymphohistiocytosis (HLH) is a clinical syndrome characterized by dysregulated immune system activation and hyperinflammation due to loss of natural killer (NK) cell and cytotoxic T lymphocyte cytolytic function leading to cytokine storm and macrophage activation [1]. HLH is rare, with an estimated annual incidence of 1 per 800 000 people, which varies by geographic population [2]. Primary HLH is the inherited form of the condition and is almost entirely isolated to early childhood, with ≤1.6% of adult cases classified as familial [3]. Secondary (reactive) HLH (sHLH) is observed in both children and adults and is an acquired form of the syndrome.
The pathogenesis of sHLH involves a predisposing disease or condition with a precipitating external trigger, primarily infection, that initiates production of the hyperinflammatory state [4,5]. Conditions that predispose to sHLH include autoimmune disease, hematologic malignancy, and organ transplant, while external triggers include infection (typically viral), autoimmune disease flare, and immunosuppression [2,4,5]. Systemic lupus erythematosus (SLE) accounts for nearly half of autoimmune-triggered sHLH [2]. When triggered by autoimmune disease, sHLH is termed macrophage activation syndrome (MAS). Although most cases of sHLH, including MAS, have an identifiable trigger, there is likely an underlying genetic predisposition to the sustained immune reaction responsible for the disease [6]. In fact, more than 40% of sHLH cases have heterozygous mutations in genes that are typically associated with homozygous mutations in primary HLH [7]. However, with some of these genes it has not been determined that heterozygous mutations result in impaired immune cell cytolytic activity [8].
Manifestations of HLH include continuous fever, lymphadenopathy, hepatosplenomegaly, mucosal bleeding, cytopenias, and other findings of inflammation and multiple-organ damage [9]. Due to the varied and nonspecific presentation of sHLH, the differential diagnosis is wide and includes sepsis, disseminated intravascular coagulation, malignancy, and autoimmune disease flare. The HScore is a useful tool in predicting the probability that a patient has reactive HLH [10]. We present the manifestation, recognition, and management of a case of macrophage activation syndrome in a patient with untreated SLE and bacteremia.
Case Report
A 43-year-old African American man with known discoid lupus presented with a 1-month history of weakness, epistaxis, shortness of breath, anorexia, and weight loss. He reported no focal weakness, fever, chest, neck or abdominal pain, headache, visual changes, hematuria or hematochezia, nausea, or vomiting. He took no medications. On examination, he was afebrile with heart rate averaging 95 beats per minute and normal respiratory rate and blood pressure. There was mucositis of the oral and nasal cavities with active mucosal bleeding. Abdominal examination revealed hepatosplenomegaly. A grade II/VI late diastolic heart murmur was noted, but the chest was clear. Laboratory findings and clinical course are included in Table 1.
Hepatitis serologies were negative and a thyroid panel and vitamin B12 and folate levels were normal. Elevated lipase was attributed to renal failure. Due to elevated hepatic enzymes, abdominal imaging was ordered in the Emergency Department (ED). CT of the abdomen and pelvis revealed mesenteric stranding suggestive of mesenteric panniculitis. Abdominal ultrasound showed hepatosplenomegaly and increased echo-genicity of bilateral renal cortices, indicative of chronic renal parenchymal disease.
The patient was admitted for a suspected lupus flare complicated by stage 3 acute kidney injury (AKI). He was started on high-dose methylprednisolone and aggressive rehydration. He received 2 rounds of plasmapheresis due to initial concern for thrombotic thrombocytopenic purpura (TTP), but no evidence of microangiopathic hemolytic anemia was found and coagulation studies were unremarkable.
Blood cultures were drawn at presentation. Piperacillin with tazobactam was started in the ED after CT revealed mesenteric panniculitis. Blood cultures on hospital day 2 returned
Our patient’s hepatosplenomegaly, leukopenia and thrombocytopenia, elevated ferritin, triglycerides, and liver enzymes, and fibrinogen less than 250 mg/dL resulted in an HScore of 225, correlating with a probability of greater than 98% of sHLH. Based on this, anakinra (an IL-1 receptor inhibitor) was initiated on hospital day 3, with remarkable clinical improvement, and reduction of his ferritin from >16 500 to 7585. Bone marrow biopsy revealed hemophagocytosis, resulting in a final HScore of 260 (Table 2, Figure 1).
Despite initial clinical improvement, his ferritin increased to >16 500. The patient’s anakinra dose was escalated and he was started on tocilizumab (an IL-6 receptor inhibitor) and low-dose etoposide. Over time, there was both clinical and laboratory improvement and the serum ferritin fell to 3307. Interestingly, an interleukin-2 (IL-2) assay completed in the hospital was normal, but a follow-up IL-2 soluble receptor level utilizing a different assay completed 1 week after discharge was elevated at 1629 (normal 222–770 U/mL).
At presentation, our patient had a creatinine elevated to 8.26 from a baseline of 0.94 in 2018. His fractional excretion of sodium was 0.3%, consistent with a prerenal etiology to his AKI. His creatinine improved from 8.26 to 4.72 on hospital day 2, and to 2.47 on hospital day 3, further indicating a significant pre-renal component to his AKI. Renal biopsy was consistent with class II lupus nephritis, but his creatinine ultimately returned to and remained within reference range. Due to his ongoing inflammatory state as well as his lupus nephritis, he received 3 days of pulsed solumedrol. By hospital day 7 his creatinine had returned to 1.05, indicating resolution of his stage 3 AKI.
Due to his positive blood cultures and diastolic heart murmur, a transthoracic echocardiogram (TTE) was performed and showed significant aortic regurgitation with normal ejection fraction. Transesophageal echocardiogram (TEE) did not reveal any vegetations or perivalvular leak.
The patient was discharged on a steroid taper, bi-weekly low-dose etoposide, and hydroxychloroquine. He continues to improve and has returned to work and normal activities.
Discussion
Despite known discoid lupus, our patient took no medications and was not established with a primary care physician. The ratio of population to primary care physicians in Oklahoma is 21.8% higher than the national ratio [11]. The percentage of Oklahomans without health insurance is also 70% higher than the national rate [11]. Even though our patient had medical insurance, he lacked access to care for his medical issues. Our patient’s lack of contact with medical care also resulted in failure to diagnose systemic lupus, rather than cutaneous-limited disease. SLE is a clinical diagnosis with no diagnostic criteria, although there are 3 different classification criteria that are often used for this purpose at the physician’s discretion [12]. The most recent classification criteria, released in 2019 by the European League Against Rheumatism and American College of Rheumatology (EULAR/ACR), use 7 clinical criteria (cumulative score 0–39) and 3 immunologic criteria (cumulative score 0–12), with a criterion being met if it has been present at any time in a patient’s disease history [12]. With a history of discoid lupus, our patient needed only positive antinuclear antibody and anti-dsDNA serologies to meet the minimum 10 points for EULAR/ACR-classified SLE, yet his lack of primary care prevented this crucial diagnosis and initiation of maintenance therapies. At presentation, our patient had a EULAR/ ACR criteria score of 35, leading to the admission diagnosis of a lupus flare. This highlights the significant possible overlap between SLE and sHLH presentations.
Sepsis may also present similarly to sHLH [13,14]. With heart rate greater than 90 beats per minute, leukopenia, and identified blood stream infection, our patient met systemic inflammatory response syndrome (SIRS) and sepsis criteria. This highlights the sensitivity of SIRS and sepsis criteria as well as a point of possible confusion in the recognition of sHLH. Discernment between the 2 conditions is complicated by the fact that approximately half of sHLH cases involve some infection [2,5], as in our patient, but most infections are viral. Notably, discontinuation of immunosuppressive therapy in favor of isolated treatment of sepsis would have been a fatal error in the care of our patient. HLH can be differentiated from sepsis by the presence of severely elevated ferritin, cytopenia of multiple lineages, and organomegaly [8]. Elevated ferritin in the setting of continued fevers and negative blood cultures may also prompt evaluation for sHLH in a patient being treated for sepsis [14]. Additionally, like our patient, sHLH can present as progressive symptoms over weeks or months rather than an acute septic picture [14].
Treatment of HLH is based on the HLH-94 and HLH-2004 protocols. The HLH-94 protocol involves 8 weeks of initial therapy with a dexamethasone taper and etoposide (twice-weekly for 2 weeks then weekly for weeks 6–8) followed by continuation therapy with dexamethasone pulses, weekly etoposide, and cyclosporine A (CSA) [15]. Intrathecal methotrexate is indicated with presence of neurologic symptoms [15]. The HLH-2004 protocol adapts HLH-94 by including CSA in the initial 8 weeks of therapy and adding intrathecal prednisolone to methotrexate with the presence of neurologic symptoms [15]. However, HLH-2004 offered no statistically significant improvement in 5-year survival over HLH-94 [15]. Due to comorbidities in adults and the numerous precipitating factors in secondary HLH, the protocols mentioned above are primarily used in pediatric populations in the treatment of familial HLH [8]. In addition to direct immunosuppression, HLH treatment in adults focuses on treating underlying conditions responsible for disease initiation and complications. With our patient, our general medicine team had significant discussion on the implications of starting aggressive immunosuppression in a patient with positive blood cultures, but ultimately, he rapidly improved with concomitant immunosuppressive and antibiotic therapies.
Despite fever being present in 96% of HLH cases [2], 36%–86% of SLE patients [16], and commonly in bacteremia, our patient presented and remained afebrile. Additionally, an IL-2 assay performed during admission was normal but an IL-2 soluble receptor level performed after discharge was elevated. Ordering IL-2 and NK cell number assays are common mistakes when evaluating for HLH [17]. Instead, IL-2 soluble receptor level and NK cell function are useful in the evaluation of HLH [17]. Even when obtained correctly, mean IL-2 soluble receptor levels in HLH are not significantly different from mean levels found in hematologic malignancy, sepsis, and rheumatologic disease [18]. IL-2 soluble receptor levels are best used for ruling out HLH, as a cutoff of 2400 U/mL yields a sensitivity of 89.2% and specificity of 38.8% [18].
Conclusions
Primary care is crucial in the recognition and management of serious chronic disease. Due to his lack of primary care, our patient possessed undiagnosed SLE that resulted in the precipitation of HLH, a severe and deadly condition. HLH has many predisposing factors and a wide array of presentations that can be difficult to distinguish from autoimmune disease flare and sepsis, among others. Our patient met the sepsis criteria and appeared to have a lupus flare, but a complete clinical picture and evaluation with tools such as the HScore led to a diagnosis of HLH when treatment for the other 2 conditions alone would have likely been fatal. Treatment of HLH is complicated and is dependent on triggering factors and concomitant comorbidities and complications. Despite having MSSA bacteremia at admission, our patient rapidly improved with immunosuppressive and antibiotic therapies.
Tables
Table 1.. Clinical course and laboratory findings.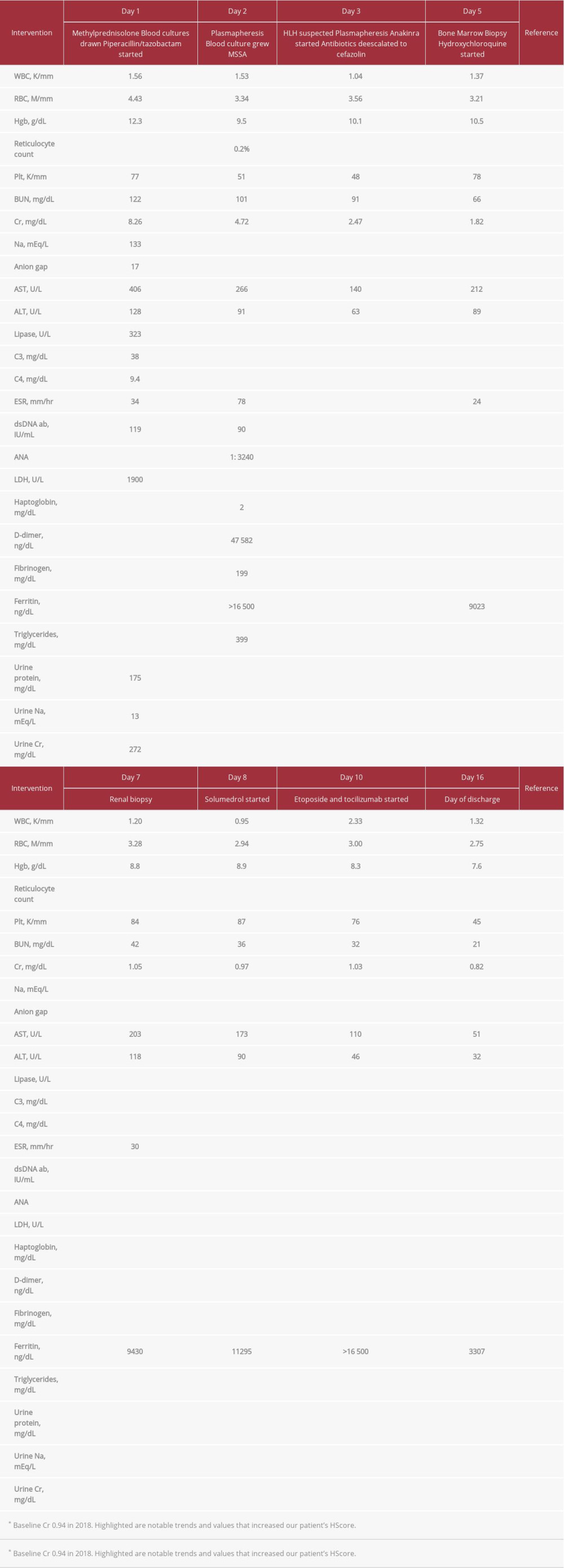 Table 2.. Shown are our patient’s presentation characteristics and how they increase his HScore, and thus his probability of having reactive HLH. Hepatosplenomegaly is based on abdominal ultrasound. Number of lineages with cytopenia is based on WBC <5 K/mm3, platelets <110 K/mm3, and hemoglobin <9.2 g/dL.
Table 2.. Shown are our patient’s presentation characteristics and how they increase his HScore, and thus his probability of having reactive HLH. Hepatosplenomegaly is based on abdominal ultrasound. Number of lineages with cytopenia is based on WBC <5 K/mm3, platelets <110 K/mm3, and hemoglobin <9.2 g/dL.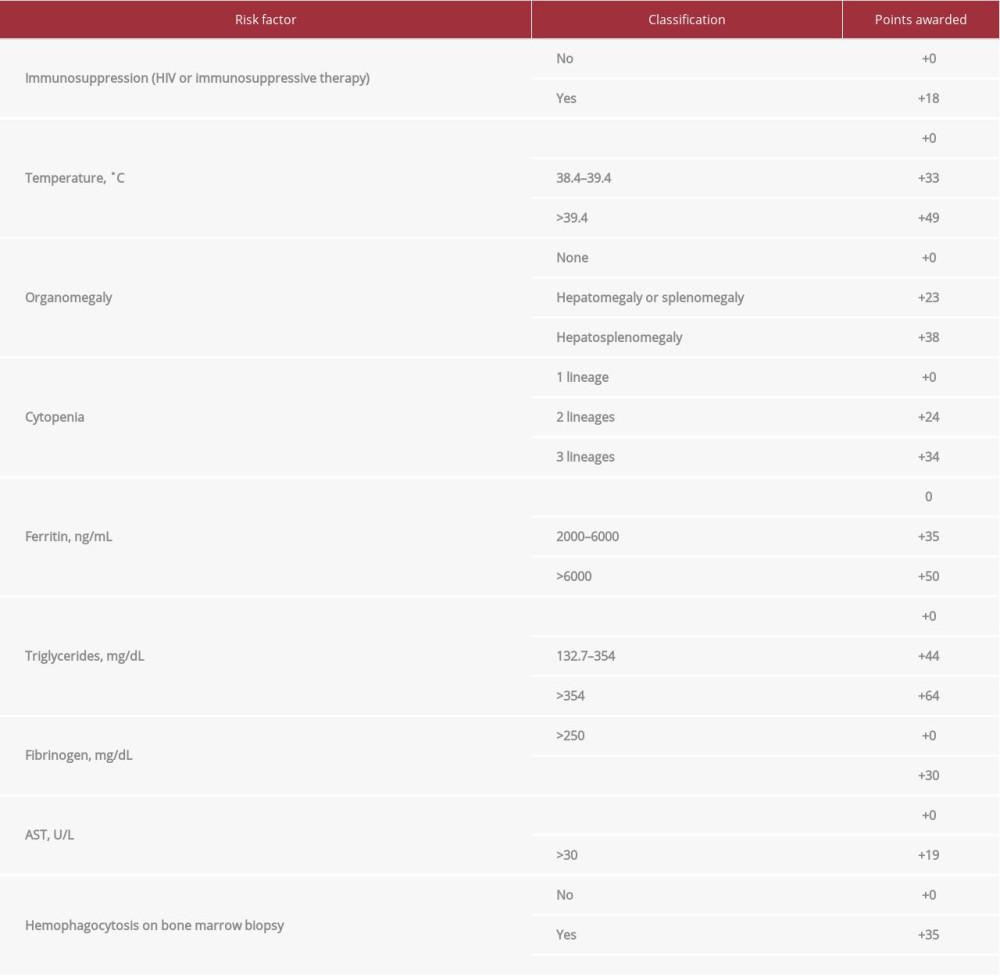
References:
1.. Carter SJ, Tattersall RS, Ramanan AV, Macrophage activation syndrome in adults: Recent advances in pathophysiology, diagnosis and treatment: Rheumatology, 2019; 58; 5-17
2.. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Adult haemophagocytic syndrome [published erratum appears in: Lancet 2014;383: 1464.]: Lancet, 2014; 383; 1503-16
3.. Wysocki CA, Comparing hemophagocytic lymphohistiocytosis in pediatric and adult patients: Curr Opin Allergy Clin Immunol, 2017; 17; 405-13
4.. Brito-Zerón P, Kostov B, Moral-Moral P, Prognostic factors of death in 151 adults with hemophagocytic syndrome: Etiopathogenically driven analysis: Mayo Clin Proc Innov Qual Outcomes, 2018; 2; 267-76
5.. Brito-Zerón P, Bosch X, Pérez-de-Lis M, Infection is the major trigger of hemophagocytic syndrome in adult patients treated with biological therapies: Semin Arthritis Rheum, 2016; 45; 391-99
6.. Griffin G, Shenoi S, Hughes GC, Hemophagocytic lymphohistiocytosis: An update on pathogenesis, diagnosis, and therapy: Best Pract Res Clin Rheumatol, 2020; 34; 101515
7.. Crayne CB, Albeituni S, Nichols KE, Cron RQ, The immunology of macrophage activation syndrome: Front Immunol, 2019; 10; 119
8.. Ponnatt TS, Lilley CM, Mirza KM, Hemophagocytic lymphohistiocytosis: Arch Pathol Lab Med, 2021 [Online ahead of print]
9.. Lerkvaleekul B, Vilaiyuk S, Macrophage activation syndrome: Early diagnosis is key: Open Access Rheumatol, 2018; 10; 117-28
10.. Fardet L, Galicier L, Lambotte O, Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome: Arthritis Rheumatol, 2014; 66; 2613-20
11.. Blomme C, Roubal A, Givens M, 2020 Oklahoma report. Univ of Wisc Pop Health Inst [online], 2020 [cited 2021 Aug 16]. Available from: URL: https://www.countyhealthrankings.org/reports/state-reports/2020-oklahoma-report
12.. Fanouriakis A, Tziolos N, Bertsias G, Boumpas DT, Update on the diagnosis and management of systemic lupus erythematosus: Ann Rheum Dis, 2021; 80; 14-25
13.. Raschke RA, Garcia-Orr R, Hemophagocytic lymphohistiocytosis: A potentially underrecognized association with systemic inflammatory response syndrome, severe sepsis, and septic shock in adults: Chest, 2011; 140; 933-38
14.. Akenroye AT, Madan N, Mohammadi F, Leider J, Hemophagocytic Lymphohistiocytosis mimics many common conditions: Case series and review of literature: Eur Ann Allergy Clin Immunol, 2017; 49; 31-41
15.. Bergsten E, Horne A, Aricó M, Confirmed efficacy of etoposide and dexamethasone in HLH treatment: Long-term results of the cooperative HLH-2004 study: Blood, 2017; 130; 2728-38
16.. Timlin H, Syed A, Haque U, Fevers in adult lupus patients [published correction appears in Cureus 2018;10: c12]: Cureus, 2018; 10; 2098
17.. Safi S, Shanbhag S, Hambley BC, Merrill SA, Systems controls are needed to reduce mistaken tests for hemophagocytic lymphohistiocytosis, results of a prospective quality-improvement cohort study: Medicine, 2021; 100; 26509
18.. Naymagon L, Tremblay D, Troy K, Mascarenhas J, Soluble interleukin-2 receptor (sIL-2r) level is a limited test for the diagnosis of adult secondary hemophagocytic lymphohistiocytosis: Eur J Haematol, 2020; 105; 255-61
Tables
Table 1.. Clinical course and laboratory findings.Table 2.. Shown are our patient’s presentation characteristics and how they increase his HScore, and thus his probability of having reactive HLH. Hepatosplenomegaly is based on abdominal ultrasound. Number of lineages with cytopenia is based on WBC <5 K/mm3, platelets <110 K/mm3, and hemoglobin <9.2 g/dL.Table 1.. Clinical course and laboratory findings.Table 2.. Shown are our patient’s presentation characteristics and how they increase his HScore, and thus his probability of having reactive HLH. Hepatosplenomegaly is based on abdominal ultrasound. Number of lineages with cytopenia is based on WBC <5 K/mm3, platelets <110 K/mm3, and hemoglobin <9.2 g/dL. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133