06 October 2022: Articles 
Pathogenic Novel Heterozygous Variant c.1076c>T p. (Ser359Phe) chr1: 120512166 in NOTCH2 Gene, Type 2 Alagille Syndrome Causing Neonatal Cholestasis: A Case Report
Unusual clinical course, Challenging differential diagnosis, Rare disease, Congenital defects / diseases
Mohammed Shahab UddinDOI: 10.12659/AJCR.935840
Am J Case Rep 2022; 23:e935840






Abstract
BACKGROUND: Alagille syndrome (ALGS) is a multisystem hereditary illness with a dominant pattern and partial penetrance. Multiple organ abnormalities can be caused by mutations in the Jagged canonical Notch ligand 1 (JAG1) gene. Notch receptor 2 (NOTCH2) gene mutations are also uncommon. ALGS is also characterized by deformed or narrowed bile ducts and is notoriously difficult to diagnose due to the wide range of symptoms and absence of unambiguous genotype-phenotype connections. Little is known about ALGS patients who have NOTCH2 mutations. We present a patient who developed progressive liver failure due to a unique pathogenic heterozygous variation of the NOTCH2 gene, c.1076c>T p. (Ser359Phe) chr1: 120512166, resulting in type 2 ALGS.
CASE REPORT: A Saudi Arabian newborn with bilateral hazy eyes, ectropion, dry ichthyic skin, normal male genitalia, and bilateral undescended testes was born at 31 weeks. Previous miscarriages, pregnancy-induced maternal cholestasis, fatty liver, or neonatal jaundice were not reported in the family history. He had developed worsening cholestatic jaundice by the third week of hospitalization. The extensive work-up for metabolic, infectious, and other relevant etiologies was negative. Following gram-negative sepsis, he died of multiorgan failure. A NOTCH2 gene mutation explained the phenotypic difference in our situation. Another intriguing observation was the presence of ichthysis and craniosynostosis in ALGS with a NOTCH2 mutation.
CONCLUSIONS: Cholestasis in newborns can be difficult to diagnose. Next-generation sequencing detects 112 copy number variants in the cholestasis gene panel blood test. More research is needed to understand why NOTCH2 mutations are relatively rare in ALGS.
Keywords: Alagille Syndrome, Cholestasis, Cholestasis, Intrahepatic, Craniosynostoses, High-Throughput Nucleotide Sequencing, JAG1 Protein, Human, Whole Exome Sequencing, Whole Genome Sequencing, Humans, Infant, Newborn, Jagged-1 Protein, Ligands, Male, Pregnancy Complications, Receptor, Notch2, Saudi Arabia
Background
Neonate cholestasis poses a diagnostic puzzle. There are over 100 disorders associated with cholestasis [1]. The best outcome depends on the identification of time-sensitive causes of cholestasis early on, such as biliary atresia [2–4], which can be treated if detected early [5–7]. Due to the complexity of the issues involved, assessing cases on a timely basis can be a challenge. Despite the best available settings, the diagnosis is usually made at 2 months of age [8]. A genetic predisposition to newborn cholestasis has been demonstrated in several studies, and family history has been found in 15% to 20% of cases [9]. The neonatal intensive care unit often has patients presenting with cholestatic jaundice brought on by genetic disorders, such as those related to bile formation, canalicular transporters, tight junction proteins, and metabolic errors [10,11]. Additionally, children with cholestasis can require liver transplantation, affecting their outlook [12–14]. Numerous genes are implicated in diseases that cause neonatal cholestasis. A pathological mutation can allow for prenatal diagnosis or outline an antenatal treatment plan in cases of life-threatening illness. Molecular diagnosis is time-consuming due to a high rate of new mutations [5]. The understanding of cholestatic diseases has greatly improved since the early 1990s. In many of these disorders that cannot be diagnosed directly by standard tests or liver biopsy, molecular diagnostics have become much more affordable and rapid through the simultaneous sequencing of multiple genes and whole genomes [15]. One in four neonates admitted to neonatal intensive care units in the United States has a genetic disorder, and those with genetic disorders spend more time in hospitals and consume more resources. Rapid whole-genome sequencing (rWGS) can improve the chances of survival for babies with genetic disorders. Furthermore, rWGS technology plays an important role in improving end-of-life care [16].
Alagille syndrome (ALGS; OMIM 118450) has many unusual and complex features, including newborn cholestasis, heart malformations, eye anomalies (posterior embryotoxon), and vertebral deformities (butterfly vertebra). In addition, a large nose, broad forehead, and pointed chin are notable facial features. Furthermore, vascular and renal abnormalities develop in a lower percentage of cases. In fact, symptoms can range from no symptoms to a life-threatening condition [17]. ALGS has a wide range of expressivity, making diagnosis difficult. A conventional diagnosis is based on the presence of interlobular bile duct paucity [18] and at least 3 of these 5 symptoms. Initially, it was found that these facial traits were not exclusive to ALGS but were linked to cholestasis. New diagnostic criteria have been developed in light of ALGS’s genetic foundation and the availability of molecular testing, which suggests that 1 main clinical characteristic and the presence of a disease-causing mutation are essential to ascertain the diagnosis [19].
Jagged canonical Notch ligand 1 (JAG1) and neurogenic locus Notch homolog protein 2 (NOTCH2) are 2 of the most common genes involved in the Notch signaling pathway (NSP) in the majority of patients with ALGS. JAG1, a gene on chromo-some 20, is responsible for almost all cases of ALGS. Only 1% to 3% of incidents are caused by a mutation in the Notch2 gene. These individuals are more likely to have renal abnormalities, although they can also less commonly have cardiac, skeletal, and facial deformities. Over 400 JAG1 mutations have been found, including deletions of whole genes, splice site modifications, mismatches, nonsense mutations, and frame shift mutations [20]. It is thought that the NOTCH2 gene on chromo-some 1p12 is responsible for Alagille syndrome 2 (ALGS2) [21]. Here, we describe a Saudi infant with a pathogenic variant in the NOTCH2 gene, c.1076c>T p. (Ser359Phe) chr1: 120512166, who presented with progressive neonatal cholestasis. It has never been reported in the literature.
Case Report
The infant was delivered by emergency cesarean delivery at 31 weeks of pregnancy, owing to abruptio placenta, and weighed 1090 g. There were Apgar values of 4 and 8 in the first and fifth minutes of life, respectively. Preceding his birth, 2 other babies perished while still within the mother’s womb. It turns out that he was the product of a multiple pregnancy. Her hypothyroidism was treated with thyroxine, but she developed gestational diabetes on a low-carbohydrate diet. Antibiotics were given to her for a group B streptococcal urine infection upon her arrival. In this case, the parents were not relative. The only living child was a 5-year-old, and he was in excellent health at the time. No noteworthy information regarding the patient’s family history was found, despite inquiries into multiple miscarriages, pregnancy-induced maternal cholestasis and fatty livers, maternal infection, and a history of newborn jaundice in the patient’s family. The family was living in Dammam, Saudi Arabia’s eastern region, and was wealthy. The infant was found to be in mild respiratory distress, with a respiration rate of 65 breaths per min and chest retraction, after the first newborn resuscitation attempt. With a CPAP machine, 6 positive end expiratory pressures and 30% inspired oxygen were used (FIO2). He was sent to the Neonatal Intensive Care Unit (NICU) for further treatment.
His physical examination showed cloudy eyes, ectropion, dry ichthyotic skin, typical male genitalia, and bilaterally undescended testicles. Other than that, he did not exhibit any signs of dysmorphia. He measured the 75th, 25th, and 25th percentiles for height, weight, and head circumference, respectively. His temperature was 36.2°C. The baby was neurologically fine. The heart rate was constant at 167 beats per min, and the pulsations were well felt. At the time, the patient was being supported by CPAP with 30% FIO2. Breath sounds from both sides of the lung were equal. There was no organomegaly in the abdomen. The remainder of the physical examination was uneventful. His blood count, electrolytes, and liver function were all normal when he was admitted to the unit. Respiratory distress syndrome ground-glass opacity was seen on the X-ray, and a minor respiratory acidosis was detected in the patient’s blood gas.
Acute respiratory distress syndrome had been diagnosed, as shown by both clinical and radiological findings. As a result, he was given a surfactant dosage and continued to receive CPAP assistance. There was a bowel movement at 12 h old. On the third day, we used trophic feeding to give the baby breast milk. He was gradually weaned from 40% to 21% FIO2 in the following weeks. On the sixth day following respiratory distress syndrome, he developed abdominal distension, tachycardia, tachypnea, thrombocytopenia, neutropenia, and a raised C-reactive protein level of 70 mg/L, and his antibiotics were escalated to meropenem and vancomycin, empirically.
Due to slight abdominal distension and leftover milk, we had to stop the tropic feeding. In the meantime, after 72 h of deterioration, the antibiotics were discontinued, since all culture results were negative, and feeding was restarted at a rate of 30 mL/kg/day. After weaning from CPAP, high-flow oxygen treatment was started in the second week of life.
Relapses of unexplained apnea occurred in his third and fourth weeks of life. He required CPAP, since his FIO2 was 30%. Each time he had a septic examination, the results were always negative. The results of brain magnetic resonance imaging (MRI) were normal. A nasogastric tube was eventually able to provide around half of his nutritional requirements, along with occasional breast milk and preterm formula. He did not have pale stools. The neonatologist recommended a neonatal cholestasis workup because of the persistence of conjugated hyperbilirubinemia (total bilirubin 69.8 umol/L=4.08 mg/dL) since the third week of hospitalization, despite negative septic workups (Table 1). Experts in the field of pediatric gastroenterology and clinical nutrition suspected a relationship between hepatitis and total parenteral nutrition (TPN). Despite an increase in nasogastric feeding volume and an adjustment in TPN, there was no change in cholestasis. Genetic studies on neonatal cholestasis, notably progressive familial intrahepatic cholestasis and ALGS, were prompted by the high frequency of genetic illnesses in the area. An X-ray of the spine, echocardiogram, and eye test all came back normal (Table 2). The skull X-ray revealed craniosynostosis (Figure 1). Recurring apnea made the hepatitis biliary scintigraphy test difficult. Hepatosplenomegaly and signs of biliary atresia were not seen on a repeat ultrasonography. There was a normal-appearing gallbladder and no signs of intrahepatic or extrahepatic biliary tree dilatation.
As a result, his albumin level began to decline over time. Bilirubin, AST, and ALT levels rose (Figures 2, 3; Table 1). Multiple albumin infusions were necessary. Due to the patient’s worsening cholestasis, despite treatment with ursodeoxycholic acid (30 mg/kg/day), an infectious etiology investigation was conducted. The results were unremarkable. (Table 2). Furthermore, the levels of TSH, T3, T4, and cortisol were not significant (Table 2). In addition, he tested negative for a wide range of metabolic disorders, including galactosemia and tyrosinemia type 1 (Table 2). No micropenis was detected during the physical examination, despite the fact that there was no history of hypoglycemia. To ultimately confirm the diagnosis, a pediatric gastroenterologist sent the whole genomic sequence to a geneticist at 8 weeks of age, ignoring the liver biopsy result, since it may not have aided a definite diagnosis.
Unfortunately, caffeine (5 mg/kg/day) and hemoglobin optimization failed to manage recurrent apnea and bradycardia, most likely due to a central origin. Furthermore, phenobarbitone was used as an empirical treatment, since portable electroencephalogram equipment were not accessible. He had severe cyanosis, apnea, and bradycardia in the 13th week of NICU treatment, necessitating 3 min of aggressive cardio-respiratory resuscitation. In addition to intubation and mechanical breathing, epinephrine was provided. Septic shock, multiorgan failure, and evidence of disseminated intravascular coagulation (DIC), acute renal damage, and capillary leak syndrome persisted over the next several days, and he remained severely sick, with a high C-reactive protein level of 223.3 mg/L. The patient died after 100 days in the hospital, despite the administration of inotropic support, high-frequency oscillating ventilation, and continuous inhaled nitric oxide treatment, due to the inability to establish an etiology for the cholestatic liver illness secondary to
The diagnostic puzzle was solved a week after his death, after we received his whole exome sequencing results. An autosomal dominant ALGS2 was caused by a pathogenic heterozygous variant c.1076c>T (Ser359Phe) chr1: 120512166 in the NOTCH2 gene.
Sampled blood with bits of genomic DNA exons and intron borders of all known human genome genes were then sequenced using the next-generation sequencing (NGS) illumina system, which uses Roche NimbleGen capture laboratory results, after approval from the parents. An average of 80-fold coverage was given to the target areas. Most areas of interest were covered by 15-fold coverage, and nearly 96% were covered by 20-fold coverage. NGS data were also used to align the HG19 genome assembly. In-house bioinformatics programs were used to call and annotate genetic variants. It was crucial to detect minor alleles with less than 1% frequency in gnomAD. Single nucleotide variants and indels were checked against internal and external databases. Even in locations with a high degree of homogeneity, known artifacts and variances were removed. Additionally, based on data from human gene mutation databases, bioinformatics prediction tools, and the current state of the literature, automatic evidence category ratings were utilized to classify variations. In databases, frequent polymorphisms and “harmless” (probable) variants were not analyzed.
An in-house quality score was used to compare the wild-type sequence (the human reference genome according to UCSC Genome Browser hg19, GRCh37) with the patient sequence presented in this research. Variants that failed the quality score were also verified using PCR amplification and conventional Sanger sequencing. As a result of quality management measures, sample identification was assured. Segregation analysis of the detected variants in the parents was recommended so we could catch de novo mutations and improve variant classification. Unfortunately, the family genetic counseling appointment had not yet been documented.
The NOTCH2 gene (OMIM *600275; chromosome 1p12) heterozygous missense variant c.1076c>T p. (Ser359Phe) (chr1: 120512166: hg19) results in an amino acid exchange. According to 8 of 10 bioinformatics in silico techniques, this variation is deleterious. To the best of our knowledge, the variant has not yet been described in the literature (HGMD 2019.2). There have been no reports of allele frequencies in the public population, and this is the first time that they have been recorded in an internal database. ALGS2 (OMIM # 610205) is characterized by hepatic bile duct paucity and cholestasis, as well as cardiac, skeletal, and ophthalmological abnormalities, and is caused by pathogenic mutations in the NOTCH2 gene (OMIM). In reality, patients’ phenotypes seem to be dependent on the information supplied. Indeed, based on the information presented, the phenotype of patients seems to support an ALGS2 diagnosis. To estimate the possibility of de novo emergence and improve variant detection, we believe that a segregation study of the discovered variation in patients is necessary. Additionally, for a deeper understanding of data interpretation, it would be beneficial to conduct a separate study of afflicted and unaffected family members. It is impossible to overestimate the value of genetic counseling for the whole family.
Indeed, any change to a protein’s structure or sequence has the potential to alter its function. Sorting Intolerant from Tolerant (SIFT) is a software that predicts how an amino acid alteration would impact protein function [22]. The SIFT score ranges from 0.0 (deleterious) to 1.0 (tolerated). The following is how the score might be interpreted: 0.05 to 0.010, variants with scores in this range are thought to be harmful. The SIFT score of 0.032 and outcome prediction were both detrimental in our scenario. Furthermore, the Mutation Taster score of 1 indicated illness producing, and the Combined Annotation Dependent Depletion tool score of 27.2 indicated pathogenic [23]. Finally, a test of the most recent functional prediction algorithms was performed, yielding a score of 1 for Deep Artificial Neural Network [24] and a score of −2.63 for Functional Analysis via Hidden Markov Models [25]. Both predicted pathogenic mutation for our case.
Discussion
We considered many differential diagnoses and ruled them out. The first concern for our newborn was cholestasis triggered by TPN, also known as TPN-associated cholestasis. The patient had recurrent bouts of apnea as well as sepsis-like symptoms, with repeated negative cultures. Since establishing a feeding regimen was difficult, he was given TPN for the majority of the duration. TPN-associated cholestasis is defined as a direct serum bilirubin concentration of more than 34 mmol/L, independent of disease-related liver enzyme abnormalities [26]. It may begin as soon as 2 weeks after TPN is administered to preterm newborns. TPN duration, birth weight, antibiotic duration, fasting, amino acids, and lipids were identified as possible exposures in a multivariate logistic regression analysis [27]. As a result, we adjusted the lipid infusion to 0.5 g/kg/day, while increasing the carbohydrate intake to 15 kcal/kg/day. Furthermore, the addition of carnitine supplementation, in addition to the treatment of ursodeoxycholic acid at 30 mg/kg/day, was unable to alleviate the patient’s cholestasis. Biliary atresia is characterized by symptoms such as conjugated hyperbilirubinemia, liver transaminases such as GGTP, and pale stool [28]. In our patient, an abdominal ultrasound was repeated, but the gallbladder was neither tiny nor nonexistent. A normal-appearing gallbladder, the lack of a triangular-cord sign, and no evidence of intrahepatic or extrahepatic biliary tree dilatation all pointed to an unlikely biliary atresia diagnosis [29].
The patient’s liver function steadily worsened as bilirubin, AST, and ALT levels rose and serum albumin fell (Figures 2, 3). A number of albumin infusions were necessary. Despite receiving ursodeoxycholic acid (30 mg/kg/day), the patient’s cholestasis worsened. A comprehensive examination found nothing wrong. All infectious etiologies, including TORCH and hepatitis A, B, and C, were negative. TSH, T3, T4, and cortisol levels were also normal (Table 2). The pertinent metabolic illnesses were evaluated and found to be negative, particularly galactosemia [30] and tyrosinemia type 1 [31]. There were no anomalies observed in a thorough metabolic examination that included blood amino acid levels, urine organic acid levels, and tandem mass spectrometry (Table 2). Fasting serum amino acid levels were raised numerous times for a variety of amino acids. These anomalies are thought to be caused by liver failure, according to pediatric metabolic and genetic experts. Despite no history of hypoglycemia, an investigation failed to confirm the diagnosis of panhypopituitarism [32]. The brain MRI was normal. At the age of 8 weeks, the patient underwent whole-exome sequencing to confirm the diagnosis.
ALGS is a multisystem illness that affects 1 in every 30 000 babies [33]. The most common clinical features of ALGS are chronic cholestasis, caused by the absence of intrahepatic bile ducts, congenital heart disease of the pulmonary outflow tract and the vasculature, butterfly vertebrae, a broad forehead, posterior embryotoxon and/or anterior segment abnormalities of the eyes, and pigmentary retinopathy. Anomalies of the basilar, carotid, and middle cerebral arteries are responsible for up to 15% of cases of minor head trauma-related intracranial bleeding [34]. A variety of structural and functional renal defects, including cysts, ureteropelvic obstructions, and renal tubular acidosis, have been described [35,36]. ALGS patients with NOTCH2 mutations and those with JAG1 mutations exhibited a great deal of diversity in the expression levels of the affected systems. The prevalence of liver malformations was universal in NOTCH2 patients, and the prevalence of ophthalmological defects and renal anomalies was identical to that of JAG1 patients. According to the study, the NOTCH2 group had a tendency toward less cardiac involvement (60.3% vs 100% in JAG1) [37]. When compared with JAG1 mutation baseline cohorts, NOTCH2 mutation probands showed a significantly lower degree of penetrance of vertebral anomalies (10%) and facial features (20%) [17]. According to one study, missense mutations account for 50% of all NOTCH2 mutations, nonsense mutations for 25%, deletions for 13%, and splice site change for 12% [37]. JAG1 and NOTCH2 mutation-related ALGS cases have recently been well described by Gilbert et al [38]. Liver biopsy results can be confusing when diagnosing ALGS. Hence, a newborn with HNF1 deficit due to a mutation in the HNF1 gene [39] or a KDM6A gene mutation has been reported to have cholestasis because of a lack of interlobular bile ducts, which is a classic sign of ALGS [40]. JAG1 mutations account for the majority of reported mutations, with NOTCH2 accounting for approximately 1% to 3% [41,42]. It has been observed that 3.2% of patients with ALGS do not have detected pathogenic mutations in either JAG1 or NOTCH2 on their genetic testing [43]. Because of the variety of changes, as well as the wide range of penetrance and clinical symptoms, diagnosing whether individuals have ALGS with NOTCH2 mutations can be challenging [42]. Cases of NOTCH2-associated ALGS are distinct from JAG1-related ALGS [44]. Over 500 cases of ALGS have been recorded since its first publication [45]. There have only been a few instances of NOTCH2 mutations in the literature. As a consequence, understanding ALGS patients with NOTCH2 mutations and their clinical presentations is crucial. Individuals with clinical signs of ALGS who do not have JAG1 mutations should be provided NOTCH2 sequencing [46]. NOTCH2 is a gene that codes for a protein known as neurogenic locus notch homolog protein 2. Signals provided by NOTCH2 receptors coupled to ligands influence many tissues throughout the body, including the heart and brain. NOTCH2 signaling [47] in a developing embryo has been shown to be critical for the formation of cells that will become part of the heart, liver, kidneys, teeth, bones, and other components [48]. Furthermore, NOTCH2 signaling defects result in a compromised immune system, tissue healing, and bone remodeling. Moreover, functional NOTCH2 is necessary for optimal kidney development, as observed in an animal trial [49]. The phenotypic profile of ALGS produced by NOTCH2 mutations may vary from that of ALGS caused by JAG1 mutations [50]. The clinical signs of NOTCH2 mutations are not as severe as those of JAG1 mutations. Recent research discovered that individuals with NOTCH2 were all impacted by liver illness. Furthermore, the incidence of ophthalmological and renal abnormalities was comparable to that of JAG1 individuals. The NOTCH2 group had less cardiac involvement than the JAG1 group (60% vs 100%, respectively). When compared to the JAG1 group, NOTCH2 probands exhibited a substantially lower incidence of vertebral malformations (10%) and facial characteristics (20%) [17]. It should also be noted that various NOTCH2 gene variants have been associated with Hajdu-Cheney syndrome [51], Serpentine fibula polycystic renal syndrome [52], and several types of cancer [53].
First, ALGS in our case was recognized using the new ALGS genetic foundation’s diagnostic criteria, which included the presence of a disease-causing mutation in the NOTCH2 gene, such as heterozygous variant c.1076c>T (Ser359Phe) chr1: 120512166, and 1 critical clinical characteristic of cholestasis [19]. Our case fits all of the criteria for newborn cholestasis. Cholestasis occurs when conjugated bilirubin (direct bili-rubin) exceeds 1 mg/dL, with total serum bilirubin less than 5 mg/dL, or when conjugated bilirubin exceeds 20% of total bilirubin over 5 mg/dL [54]. According to one study, the criterion for raised conjugated bilirubin is more than 0.5 mg/dL, while the cutoff for elevated direct bilirubin is greater than 2 mg/dL in the first 14 days after delivery [55]. Total bilirubin was high (4.08 mg/dL, or 69.8 umol/L), and direct bilirubin was 1.27 mg/dL, an increase of more than 20% since the start of the third week.
Second, because of the diversity of alterations, as well as the broad range of penetrance and clinical symptoms, determining whether someone has ALGS syndrome with NOTCH2 mutations can be difficult. Our patient with ALGS2 had no additional organ problems. Third, as 1 of 3 triplets, this infant was delivered prematurely. We emphasize that these other 2 infants perished intrauterine while still in the pregnancy. Both of them were quite likely to have perished as a result of genetic flaws. Findings have shown that mice with homozygous NOTCH2 mutations have kidney problems as fetuses, as well as hypoplastic cardiac malformations, and are more likely to die intrauterine as a result [56]. However, none of these assumptions are guaranteed to be correct. Fourth, we believe it is important to mention that another notable finding in our instance was craniosynostosis, which has not before been associated with NOTCH2 mutations. There had previously been reports of craniosynostosis in individuals with ALGS and JAG1 mutations, but no reports of NOTCH2 mutations. Several genetic studies have shown a link between the Twist Family BHLH Transcription Factor 1 (TWIST1) gene and the NOTCH2 gene. TWIST1 has been demonstrated to positively regulate JAG1 at mesenchymal sutures, while JAG1 inhibits NOTCH2 in the mesenchyme. JAG1/NOTCH is essential for the early specification of sutural cells as well as the development of the coronal suture’s border between the osteogenic and non-osteogenic compartments. We hypothesize that the novel NOTCH2 mutation was 1 of the factors affecting the unique fused suture seen in our case [57].
The phenotypic variation that we observed in our case was due to a novel mutation. In addition to the paucity of typical phenotypic features of ALGS, additional features, such as ichthyosis and craniosynostosis, have never been reported for ALGS with a NOTCH2 mutation. A test to detect single nucleotide and copy number variants in 112 genes that are related to neonatal cholestasis is called the Cholestasis Gene Panel. Next-generation sequencing technology [58] is used to perform this test. Detection of a pathogenic variant in cholestasis may provide helpful information with respect to diagnosis, prognosis, clinical management, genetic counseling, and familial screening.
Conclusions
Many of the etiologies shown to be involved in the development of newborn cholestasis are genetic in nature. The challenge of determining the priority of diagnostic investigations in infants is especially difficult since their clinical presentations are so similar. A multigene panel would be able to provide a speedy and comprehensive analysis.
Figures
References:
1.. Feldman AG, Sokol RJ, Recent developments in diagnostics and treatment of neonatal cholestasis: Semin Pediatr Surg, 2020; 29(4); 150945
2.. Wadhwani SI, Turmelle YP, Nagy R, Prolonged neonatal jaundice and the diagnosis of biliary atresia: A single-center analysis of trends in age at diagnosis and outcomes: Pediatrics, 2008; 121(5); e1438-e40
3.. Sokol RJ, Mac C, Narkewicz MR, Karrer FM, Pathogenesis and outcome of biliary atresia: Current concepts: J Pediatr Gastroenterol Nutr, 2003; 37(1); 4-21
4.. Serinet MO, Wildhaber BE, Broué P, Impact of age at Kasai operation on its results in late childhood and adolescence: A rational basis for biliary atresia screening: Pediatrics, 2009; 123(5); 1280-86
5.. Girard M, Lacaille F, Diagnosis of neonatal cholestasis: Ann Nestle, 2008; 66(3); 109-20
6.. Mckiernan PJ, Neonatal cholestasis: Semin Neonatol, 2002; 7(2); 153-65
7.. Götze T, Blessing H, Grillhösl C, Neonatal cholestasis – differential diagnoses, current diagnostic procedures, and treatment: Front Pediatr, 2015; 3; 43
8.. Pandita A, Gupta V, Gupta G, Neonatal cholestasis: A Pandora’s box: Clin Med Insights Pediatr, 2018; 12; 117955651880541
9.. Benchimol EI, Walsh CM, Ling SC, Early diagnosis of neonatal cholestatic jaundice: Test at 2 weeks: Can Fam Physician, 2009; 55(12); 1184-92
10.. Feldman AG, Sokol RJ, Recent developments in diagnostics and treatment of neonatal cholestasis: Semin Pediatr Surg, 2020; 29(4); 150945
11.. Togawa T, Sugiura T, Ito K, Molecular genetic dissection and neonatal/infantile intrahepatic cholestasis using targeted next-generation sequencing: J Pediatr, 2016; 171; 171-177.e4
12.. Moini M, Mistry P, Schilsky ML, Liver transplantation for inherited metabolic disorders of the liver: Curr Opin Organ Transplant, 2010; 15(3); 269-76
13.. Schöning W, Schmeding M, Ulmer F, Liver transplantation for patients with cholestatic liver diseases: Visz Gastrointest Med Surg, 2015; 31(3); 194-98
14.. Mehl A, Bohorquez H, Serrano M-S, Liver transplantation and the management of progressive familial intrahepatic cholestasis in children: World J Transplant, 2016; 6(2); 278
15.. Feldman AG, Sokol RJ, Neonatal cholestasis: Emerging molecular diagnostics and potential novel therapeutics: Nat Rev Gastroenterol Hepatol, 2019; 16(6); 346-60
16.. Farnaes L, Hildreth A, Sweeney NM, Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization: NPJ Genom Med, 2018; 3; 10
17.. Kamath BM, Hutchinson A, Bauer R, 26 Notch2 mutations in Alagille syndrome: J Hepatol, 2011; 54(2); S12-13
18.. Hata K, Eto T, [Alagille syndrome.]: Ryoikibetsu Shokogun Shirizu, 1995(7); 523-26 [in Japanese]
19.. Lin HC, Hoang P Le, Hutchinson A, Alagille syndrome in a Vietnamese cohort: Mutation analysis and assessment of facial features: Am J Med Genet A, 2013(5); 1005-13
20.. Zhang E, Xu Y, Yu Y, JAG1 loss-of-function mutations contributed to Alagille syndrome in two Chinese families: Mol Med Rep, 2018; 18(2); 2356-64
21.. Kim KY, Kim TH, Seong MW, Mutation spectrum and biochemical features in infants with neonatal Dubin-Johnson syndrome: BMC Pediatr, 2020; 20(1); 1-7
22.. Ng PC, Henikoff S, SIFT: Predicting amino acid changes that affect protein function: Nucleic Acids Res, 2003; 31(13); 3812-14
23.. Nakagomi H, Mochizuki H, Inoue M, Combined annotation-dependent depletion score for BRCA1/2 variants in patients with breast and/or ovarian cancer: Cancer Sci, 2018; 109(2); 453-61
24.. Quang D, Chen Y, Xie X, DANN: A deep learning approach for annotating the pathogenicity of genetic variants: Bioinformatics, 2015; 31(5); 761-63
25.. Shihab HA, Rogers MF, Gough J, An integrative approach to predicting the functional effects of non-coding and coding sequence variation: Bioinformatics, 2015; 31(10); 1536-43
26.. Jolin-Dahel K, Ferretti E, Montiveros C, Parenteral nutrition-induced cholestasis in neonates: Where does the problem lie?: Gastroenterol Res Pract, 2013; 2013; 163632
27.. Alkharfy TM, Ba-Abbad R, Hadi A, Total parenteral nutrition-associated cholestasis and risk factors in preterm infants: Saudi J Gastroenterol, 2014; 20(5); 293-96
28.. Chardot C, Biliary atresia: Orphanet J Rare Dis, 2006; 1(1); 28
29.. Lee SM, Cheon JE, Choi YH, Ultrasonographic diagnosis of biliary atresia based on a decision-making tree model: Korean J Radiol, 2015; 16(6); 1364-72
30.. Kavehmanesh Z, Torkaman M, Beiraghdar F, A case of classic galactosemia manifesting as neonatal early and profound indirect hyperbilirubinemia: Turkish Arch Pediatr, 2020; 55(3); 316-19
31.. Ghazy RM, Khedr MA, Neonatal cholestasis: Recent insights: Egyptian Pediatric Association Gazette, 2019; 3; 67
32.. Hodges S, Buckler JMH, Neonatal cholestasis and hypopituitarism: Arch Dis Child, 1984; 59(12); 1200
33.. Micaglio E, Andronache AA, Carrera P, Novel JAG1 deletion variant in patient with atypical alagille syndrome: Int J Mol Sci, 2019; 20(24); 6247
34.. Kamath BM, Spinner NB, Emerick KM, Vascular anomalies in Alagille syndrome: A significant cause of morbidity and mortality: Circulation, 2004; 109(11); 1354-58
35.. Turnpenny PD, Ellard S, Alagille syndrome: Pathogenesis, diagnosis and management: Eur J Hum Genet, 2012; 20(3); 251-57
36.. Emerick KM, Rand EB, Goldmuntz E, Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis: Hepatology, 1999; 29(3); 822-29
37.. Leonard LD, Chao G, Baker A, Clinical utility gene card for: Alagille syndrome (ALGS): Eur J Hum Genet, 2014; 22(3); 436
38.. Gilbert MA, Bauer RC, Rajagopalan R, Alagille syndrome mutation update: Comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification: Hum Mutat, 2019; 40(12); 2197-220
39.. Pinon M, Carboni M, Colavito D, Not only Alagille syndrome. Syndromic paucity of interlobular bile ducts secondary to HNF1β deficiency: A case report and literature review: Ital J Pediatr, 2019; 45(1); 27
40.. Masui D, Fukahori S, Mizuochi T, Cystic biliary atresia with paucity of bile ducts and gene mutation in KDM6A: A case report: Surg Case Reports, 2019; 5(1); 132
41.. Turnpenny PD, Ellard S, Alagille syndrome: Pathogenesis, diagnosis and management: Eur J Hum Genet, 2012; 20(3); 251-57
42.. Shaul E, Kogan-Liberman D, Schuckalo S, Novel mutations in NOTCH2 gene in infants with neonatal cholestasis: Pediatr Rep, 2019; 11(3); 53-55
43.. Ayoub MD, Kamath BM, Alagille syndrome: Diagnostic challenges and advances in management: Diagnostics, 2020; 10(11); 907
44.. Kamath BM, Bauer RC, Loomes KM, NOTCH2 mutations in Alagille syndrome: J Med Genet, 2012; 49(2); 138-44
45.. Pati GK, Singh A, Nath P, A 10-year-old child presenting with syndromic paucity of bile ducts (Alagille syndrome): A case report: J Med Case Rep, 2016; 10(1); 342
46.. Kamath BM, Spinner NB, Rosenblum ND, Renal involvement and the role of Notch signalling in Alagille syndrome: Nat Rev Nephrol, 2013; 9(7); 409-18
47.. Krantz ID, Piccoli DA, Spinner NB, Alagille syndrome: J Med Genet, 1997; 34(2); 152-57
48.. Andersson ER, Chivukula IV, Hankeova S, Mouse model of Alagille syndrome and mechanisms of Jagged1 missense mutations: Gastroenterology, 2018; 154(4); 1080-95
49.. Mukherjee M, Fogarty E, Janga M, Surendran K, Notch signaling in kidney development, maintenance, and disease: Biomolecules, 2019; 9(11); 692
50.. Gilbert MA, Spinner NB, Alagille syndrome: Genetics and functional models: Curr Pathobiol Rep, 2017; 5(3); 233-41
51.. Canalis E, Zanotti S, Hajdu-Cheney syndrome: A review: Orphanet J Rare Dis, 2014; 9; 200
52.. Gray MJ, Kim CA, Bertola DR, Serpentine fibula polycystic kidney syndrome is part of the phenotypic spectrum of Hajdu-Cheney syndrome: Eur J Hum Genet, 2012; 20(1); 122-24
53.. Arcaini L, Rossi D, Lucioni M, The notch pathway is recurrently mutated in diffuse large B-Cell lymphoma associated with hepatitis c virus infection: Haematologica, 2015; 100(2); 246-52
54.. Feldman AG, Sokol RJ, Neonatal cholestasis: Neoreviews, 2013; 14(2); e63
55.. Davis AR, Rosenthal P, Escobar GJ, Newman TB, Interpreting conjugated bilirubin levels in newborns: J Pediatr, 2011; 158(4); 562-55
56.. McCright B, Gao X, Shen L, Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation: Development, 2001; 128(4); 491-502
57.. Chaisrisawadisuk S, Vatanavicharn N, Praphanphoj V, Bilateral squamosal synostosis: Unusual presentation of chromosome 1p12–1p13.3 deletion. Illustrative case: J Neurosurg Case Lessons, 2021; 1(3); 10-14
58.. Lipiński P, Ciara E, Jurkiewicz D, Targeted next-generation sequencing in diagnostic approach to monogenic cholestatic liver disorders – single-center experience: Front Pediatr, 2020; 8; 414
Figures
Tables
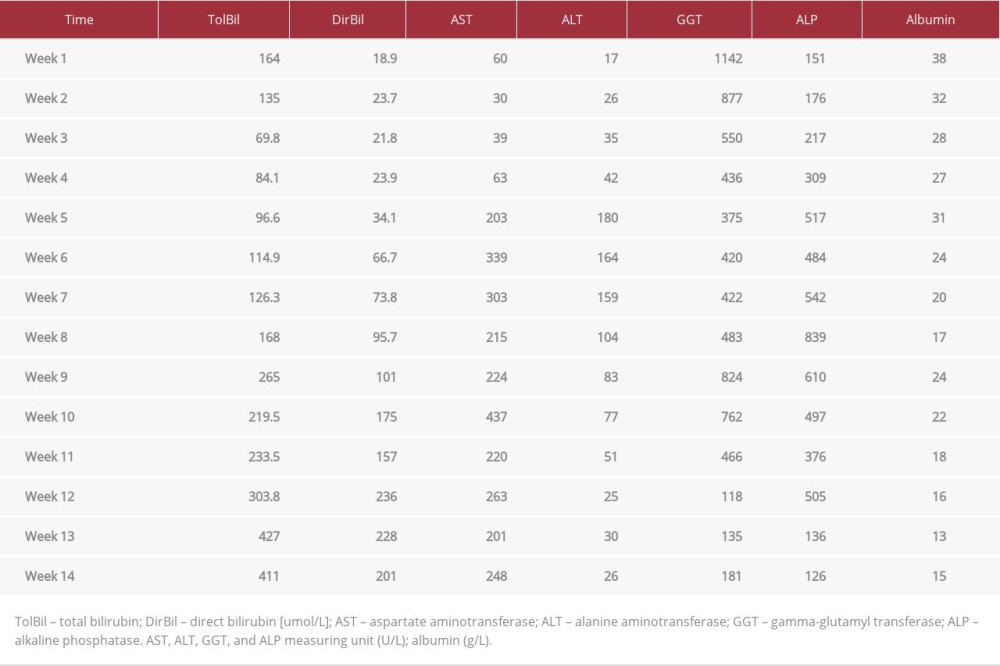 Table 1.. Serum bilirubin level during hospitalization from the week 1 to week 14.
Table 1.. Serum bilirubin level during hospitalization from the week 1 to week 14. Table 2.. Laboratory workup for neonatal cholestasis.
Table 2.. Laboratory workup for neonatal cholestasis.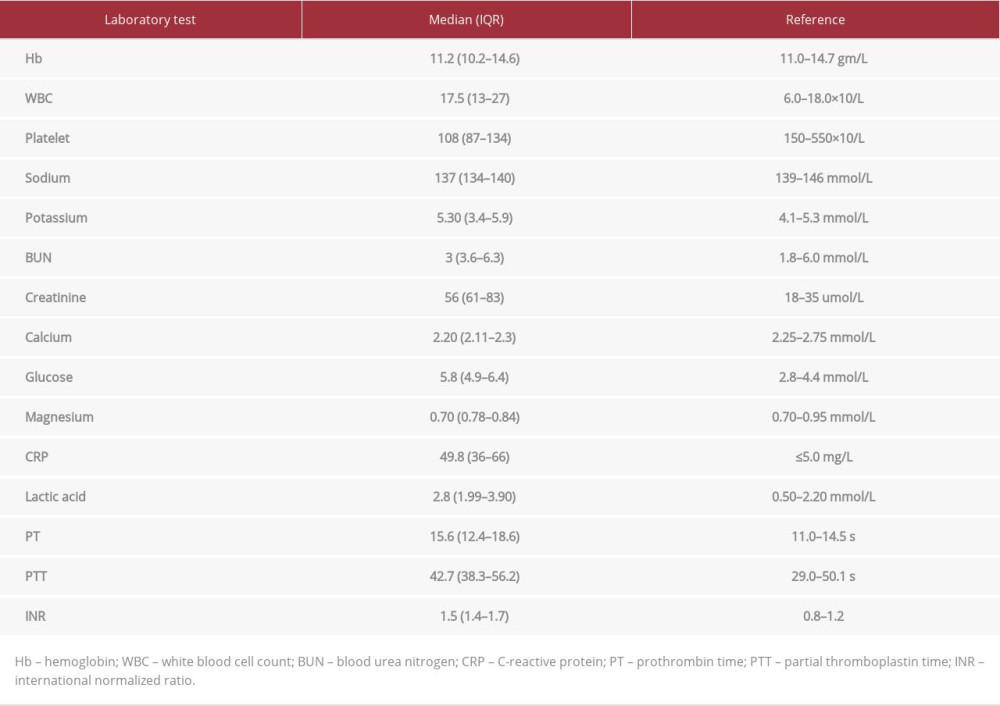 Table 3.. Laboratory findings during hospitalization.Table 1.. Serum bilirubin level during hospitalization from the week 1 to week 14.Table 2.. Laboratory workup for neonatal cholestasis.Table 3.. Laboratory findings during hospitalization.
Table 3.. Laboratory findings during hospitalization.Table 1.. Serum bilirubin level during hospitalization from the week 1 to week 14.Table 2.. Laboratory workup for neonatal cholestasis.Table 3.. Laboratory findings during hospitalization. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report  22,759,844
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133