31 August 2022: Articles 
Encephalitis with Extensive Cortical Brain Magnetic Resonance Imaging Changes Secondary to Myelin Oligodendrocyte Glycoprotein Antibody Disease
Challenging differential diagnosis, Rare disease
Vivek Satyasi1BEF*, Gayatra Mainali2AEF, Jahnavi Chatterjee2EF, Sita Paudel2AEFDOI: 10.12659/AJCR.936361
Am J Case Rep 2022; 23:e936361






Abstract
BACKGROUND: The relatively new autoimmune disorder, anti-myelin oligodendrocyte glycoprotein (MOG) disease is particularly interesting because of its broad range of presentations. This entity’s appearance on magnetic resonance imaging (MRI) of the brain often makes identifying this disease a challenging process. Younger patients tend to present with an acute disseminated encephalomyelitis picture, with encephalopathy and multifocal neurological signs, while older patients are more likely to present with optic neuritis. We, however, report an atypical case of a patient who presented with encephalopathy, seizures, and significant cortical and subcortical gray matter involvement and was found to have anti-MOG positivity in serum.
CASE REPORT: A 17-month-old previously healthy boy presented to Emergency Department with fever, lethargy, vomiting, and left-sided weakness. Eventually, he required intubation due to a prolonged seizure. Continuous electroencephalogram captured several focal seizures, and MRI of the brain showed cortical and subcortical T2 hyperintensities. After extensive laboratory evaluation, he tested positive for anti-MOG antibody. He was empirically started on high-dose intravenous pulse methylprednisolone, followed by plasma exchange, given the poor response to the intravenous steroids. At the 5-month follow-up, the results of the neurological examination had dramatically improved, and MRI findings had largely resolved.
CONCLUSIONS: This case highlights the importance of suspecting anti-MOG antibody-mediated encephalitis, even while ruling out infectious etiologies, in children presenting with encephalopathy, seizures and MRI abnormalities. Prompt recognition would allow for less delay in treatment and hopefully improve prognosis.
Keywords: Encephalitis, Epilepsy, Focal, MOG Protein, Human, Pediatrics, Autoantibodies, Humans, Magnetic Resonance Imaging, Male, Myelin-Oligodendrocyte Glycoprotein, Seizures
Background
Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) is a relatively novel entity among autoimmune encephalitides. MOGAD is of particular interest owing to its broad range of presentation. The MOG antigen is a central nervous system-specific glycoprotein that is located on the surface of myelin and oligodendrocytes. Its role is not clearly known, but it may function as a receptor or a cell adhesion molecule [1]. Even though the MOG antigen is a quantitively minor component, it is highly immunogenic. Brain biopsy studies point to activated complement deposition in perivascular spaces and in active demyelinating lesions, along with macrophages, CD4-predominant T cells, and few CD20- and CD79a-positive B cells [1–3]. Some of these studies showed a predominance of perivenous and confluent white matter lesions and intracortical lesions, while other cases showed mainly cortico-medullary junction lesions. Anti-MOG antibodies can be associated with several pediatric demyelinating syndromes, including acute disseminated encephalomyelitis (ADEM), optic neuritis, neuromyelitis optica-like disease, and multiple sclerosis [4,5]. Rarely, this autoimmune condition can present as a form of encephalitis that clinically and radiologically can resemble that of an infectious or neoplastic etiology [6,7]. European studies show an overall prevalence of this disease to be 1.6 per million, with a predominance in White patients [8,9]. These studies also show that the sex ratio of MOGAD is close to 1: 1, with a higher rate of seropositivity among children, although the disease phenotype can vary between children and adults [9–11]. An Australian study with a cohort of 59 patients showed that, among children with sero-positive MOGAD, most presented with ADEM, whereas adults mostly presented with unilateral or bilateral optic neuritis [12].
We describe a 17-month-old boy who presented with encephalopathy and seizures, with extensive cortical and subcortical magnetic resonance imaging (MRI) changes in the brain, secondary to anti-MOG antibodies. This case primarily serves to report on an uncommon presentation of anti-MOGAD as well as to provide commentary on outcomes with the treatment administered to this patient.
Case Report
A 17-month-old previously healthy and developmentally normal boy presented to the Emergency Department with 3-day history of fever, increasing lethargy, vomiting, and left-sided weakness. In the Emergency Department, the head computed tomography (CT) scan was normal. After the scan, he had a bilateral tonic-clonic seizure lasting longer than 10 min. He was given intravenous (i.v.) benzodiazepines, along with empiric antibiotics. He was intubated due to respiratory failure and was transferred to the Intensive Care Unit for further treatment. His condition on examination after intubation was limited by the sedatives but was notable for weakness, evidenced by decreased withdrawal to painful stimuli in the left upper and lower extremities.
Serial lumbar puncture revealed persistently elevated opening pressures and lymphocytic predominance, concerning for an aseptic meningitis; however, repeated studies failed to demonstrate the presence of viral nucleic acid via polymerase chain reaction (PCR) testing, including herpes simplex virus (Table 1). A serum toxicology screen was negative for any drug or toxic agent, with the exception of benzodiazepines, which he had received in the hospital. An infectious workup did reveal a positive respiratory viral panel for enterovirus/rhinovirus, but further testing showed that there was no detectable entero-virus in the serum or cerebrospinal fluid (CSF) by PCR testing. Our team had even ordered a meningitis/encephalitis panel by PCR, which is a broad panel designed to detect several different viral, bacterial, and even fungal pathogens in the CSF. While this test returned back as negative, it did require almost a week to process.
While undergoing these tests, continuous electroencephalogram (EEG) was started and demonstrated focal subclinical seizures involving the right hemisphere, which occurred periodically throughout the first 24 h and responded well to levetiracetam and phenobarbital. MRI of the brain with and without contrast showed bilateral and multifocal areas of restricted diffusion localized to the cortex. Fluid-attenuated inversion recovery (FLAIR) hyperintensities in the same regions and in the right thalamus were also shown, raising suspicion for an autoimmune encephalitis, especially given the lack of evidence in the history and laboratory studies to suggest another toxic, metabolic, or other infectious etiology (Figures 1, 2A, 2B). A genetic etiology was also briefly considered, but because this was a subacute to acute presentation and a first occurrence, further investigation into this etiology was deferred. A frequent test that is used when suspicion for autoimmune or para-infectious processes are suspected is the autoimmune encephalopathy panel, which is a panel of tests geared to detect antibodies to different antigens that are frequently associated with neurological disease. Our team had ordered this panel in both the serum and CSF, as is the common practice to increase sensitivity, and sent it to the Mayo Clinic in Rochester, Minnesota, where these tests are analyzed. Additionally, anti-MOG antibody testing was ordered mainly owing to its known association with a more common syndrome in our patient’s age group, known as ADEM [4,5]. While it has different imaging characteristics, more confluent hyperintensities on FLAIR MRI imaging with white matter involvement, it still falls on the same spectrum of para-infectious encephalitis that was being considered for our patient.
At this point in the patient’s case, after we had thoroughly evaluated for an infectious etiology and had not had much evidence to keep this etiology high on the differential diagnosis, a high-dose intravenous (i.v.) methylprednisolone course was started for 5 days, followed by 6 weeks of oral prednisone taper, mainly to treat a presumed autoimmune or para-infectious encephalitis.
Within 3 days of starting the steroid treatment, the patient began to improve in regard to his encephalopathy, lack of seizures, and epileptiform activity on the EEG and left-sided hemiparesis; however, because of the lesion burden in MRI and paucity of data in regard to treatment of autoimmune encephalitis, the decision was made to start plasmapheresis for 5 sessions after finishing 5 days of steroids on day 7 of hospitalization. Throughout the course of his treatment, he continuously improved, despite a complication of central line misplacement in the subclavian artery, which required surgical correction. After having recovered from this complication, by day 18 of hospitalization, his findings on examination dramatically improved, with only mild residual left-sided hemiparesis present, but he was able to walk and interact with his surroundings. All of his serological and CSF testing was unremarkable, except for positive serum anti-MOG antibody.
The patient spent the next month in the hospital continuing his recovery and receiving speech, physical, and occupational therapy. He was discharged a month later to the rehabilitation hospital. Two months after his discharge, he was evaluated in the outpatient Pediatric Neurology clinic and had greatly improved and returned almost back to baseline, without significant residual weakness or recurrent seizures. At 5 months after presentation, a repeat MRI of the brain showed resolution of most, if not all, FLAIR and diffusion-weighted hyperintensities, albeit with parenchymal volume loss (Figures 3, 4). Additionally, a repeat EEG in 5 months also showed improvement.
Discussion
This case is an example of a young pediatric patient with new-onset seizures, secondary to a subset of MOGAD, called FLAIR, with imaging showing cortical hyperintense lesions in MOG-associated encephalitis with seizures, otherwise referred to as “FLAMES”. This is a rare but reported entity, characterized typically by seizures, headache, fever, and the classic imaging findings of cortical hyperintensities, which are typically uni-lateral, but in a few cases, as demonstrated in our patient, can be bilateral [6,7].
A previous case report by Budhram et al documented cases in patients ranging from 11 to 46 years of age, and another case report by Ogawa et al described patients with an age range of 23 to 39 years [8]. A case report focused on pediatric MOG-associated encephalitis reported an age range of 3 to 16 years [9]. Our case is unique in that the patient was very young, 17 months, at the time of presentation, highlighting the need to recognize this disease entity as a possible etiology even in younger children.
The typical course and progression of the disease includes seizures, fever, and encephalopathy, usually preceded by an antecedent illness. Patients can initially be treated with empiric antibiotics owing to suspicion for infectious encephalitis; however, failure to respond, a lack of convincing evidence on laboratory testing in both serum and CSF, and a negative MRI should also raise concern for an antibody-mediated encephalitis [8,10,11]. If patients are given timely i.v. steroids, such as methylprednisolone, the prognosis is quite good, with near functional independence [6]. Most of the patients in the case reports and series received i.v. steroids or i.v. immune globulin [6]. Rarely, patients required disease-modifying therapy, such as rituximab, but there is no previously reported use of plasma exchange, making our case even more unique [11–14]. Plasma exchange has been used for demyelinating presentations of MOGAD, but not much for encephalitis. The exact mechanisms of the effect of these treatments are unclear, but the most well-accepted idea is that these medication regimens attenuate the overall immune response and promote an anti-inflammatory state [1–3,11].
FLAMES can result in unilateral and bilateral cortical changes similar to what was observed in our patient (Figures 1, 2A, 2B). It is important to note, however, that while these findings are distinct, they are not specific for FLAMES or anti-MOG encephalitis. It is key to rule out other causes, such as postictal edema, subarachnoid hemorrhage, infectious etiologies, and other toxidromes, which was imperative in our case before advancing to treating, specifically for an autoimmune encephalitis [1].
Given the short time frame from discharge, it is unclear whether our patient will have a recurrence in symptoms, or how long he will need anti-seizure medications, but 4 of 10 pediatric patients in a small case series had at least 1 relapse, and 1 patient had up to 3 relapses [6]. These patients ended up receiving disease-modifying therapy, such as rituximab. It remains to be seen if the decision to treat with plasma exchange would at all change the risk of relapse, but with close monitoring in the outpatient setting, appropriate action can be taken.
Regarding MOG antibody testing, our patient’s titer was 1: 100 at the onset, but the average in the pediatric case report was 1: 640, compared with <1: 160 at approximately 11 months following the acute illness [10,12,13]. While our patient did not receive repeat anti-MOG antibody testing, as a part of future follow-up visits, testing serum antibody levels may help assess the likelihood of relapse, as those with relapse were more commonly found to have higher titers than those with monophasic illness [10,13].
Conclusions
This case highlights the importance of considering MOG-associated encephalitis as a possibility for new-onset seizures, especially given characteristic MRI findings of cortical hyperintensities without white matter involvement and a lack of infectious etiologies. While this case report primarily concerns a single patient, the choice of treatment and eventual great functional outcome should spark discussion and consideration for prospective trials to determine the best treatment strategy for patients with similar presentations. This knowledge would help prioritize the prompt treatment and close monitoring of patients after their initial illness to monitor for relapse and the need for further disease-modifying therapy.
Figures
References:
1.. Fernandez-Carbonell C, Vargas-Lowy D, Musallam A, Clinical and MRI phenotype of children with MOG antibodies: Mult Scler, 2016; 22(2); 174-84
2.. Johns TG, Bernard CC, The structure and function of myelin oligodendrocyte glycoprotein: J Neurochem, 1999; 72; 1-9
3.. Takai Y, Misu T, Kaneko K, Myelin oligodendrocyte glycoprotein antibody-associated disease: An immunopathological study: Brain, 2020; 143(5); 1431-46
4.. Baumann M, Sahin K, Lechner C, Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein: J Neurol Neurosurg Psychiatry, 2015; 86(3); 265-72
5.. Marino A, Canonico F, Pinzani RM, Autoimmune rhomboencephalitis: A pediatric case report: Turk Pediatri Ars, 2020; 55(4); 449-52
6.. Budhram A, Mirian A, Le C, Hosseini-Moghaddam SM, Unilateral cortical FLAIR-hyperintense lesions in anti-MOG-associated encephalitis with seizures (FLAMES): Characterization of a distinct clinico-radiographic syndrome: J Neurol, 2019; 266(10); 2481-87
7.. Jain K, Cherian A, Divya KP, FLAMES: A novel burning entity in MOG IgG associated disease: Mult Scler Relat Disord, 2021; 49; 102759
8.. Ogawa R, Nakashima I, Takahashi T, MOG antibody – positive, benign, unilateral, cerebral cortical encephalitis with epilepsy: Neurol Neuroimmunol Neuroinflamm, 2017; 4(2); e322
9.. Wegener-Panzer A, Cleaveland R, Wendel EM, Clinical and imaging features of children with autoimmune encephalitis and MOG antibodies: Neurol Neuroimmunol Neuroinflamm, 2020; 7(4); e731
10.. Hennes EM, Baumann M, Schanda K, Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome: Neurology, 2017; 89(9); 900-8
11.. Jurynczyk M, Messina S, Woodhall MR, Clinical presentation and prognosis in MOG-antibody disease: A UK study: Brain, 2017; 140(12); 3128-38
12.. Cobo-Calvo A, Ruiz A, Maillart E, Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: The MOGADOR study: Neurology, 2018; 90(21); 1858-69
13.. de Mol CL, Wong Y, van Pelt ED, The clinical spectrum and incidence of anti-MOG-associated acquired demyelinating syndromes in children and adults: Mult Scler, 2020; 26(7); 806-14
14.. Ramanathan S, Mohammad S, Tantsis E, Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination: J Neurol Neurosurg Psychiatry, 2018; 89(2); 127-37
Figures
Tables
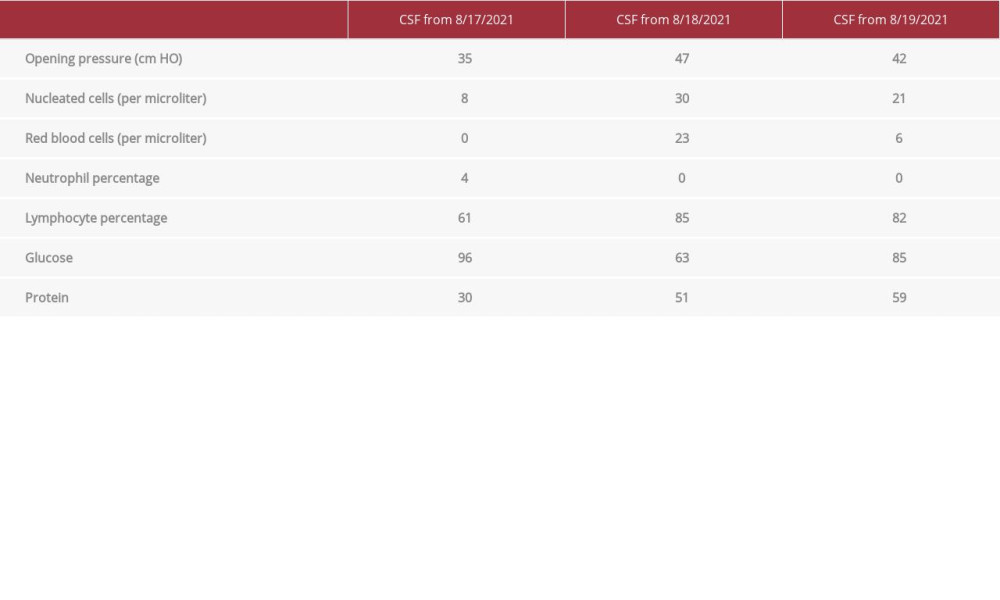 Table 1.. Results of 3 cerebral spinal fluid (CSF) samples collected over 3 days. Initially, the nucleated cells were only mildly elevated, but in the next 2 days, the nucleated cells progressively rose. There was a persistent lymphocytic predominance and mild protein elevation, but glucose levels were within the reference range. Opening pressure continued to be elevated.Table 1.. Results of 3 cerebral spinal fluid (CSF) samples collected over 3 days. Initially, the nucleated cells were only mildly elevated, but in the next 2 days, the nucleated cells progressively rose. There was a persistent lymphocytic predominance and mild protein elevation, but glucose levels were within the reference range. Opening pressure continued to be elevated.
Table 1.. Results of 3 cerebral spinal fluid (CSF) samples collected over 3 days. Initially, the nucleated cells were only mildly elevated, but in the next 2 days, the nucleated cells progressively rose. There was a persistent lymphocytic predominance and mild protein elevation, but glucose levels were within the reference range. Opening pressure continued to be elevated.Table 1.. Results of 3 cerebral spinal fluid (CSF) samples collected over 3 days. Initially, the nucleated cells were only mildly elevated, but in the next 2 days, the nucleated cells progressively rose. There was a persistent lymphocytic predominance and mild protein elevation, but glucose levels were within the reference range. Opening pressure continued to be elevated. In Press
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133