28 October 2022: Articles 
Novel Complement Factor B Gene Mutation Identified in a Kidney Transplant Recipient with a Shiga Toxin-Triggered Episode of Thrombotic Microangiopathy
Rare disease
Joanna Korzycka1BCDEF*, Ewa Pawłowicz-SzlarskaDOI: 10.12659/AJCR.936565
Am J Case Rep 2022; 23:e936565






Abstract
BACKGROUND: Atypical hemolytic uremic syndrome (aHUS) is an ultra-rare disorder characterized by over-activation and dysregulation of the alternative complement pathway. The clinical presentation of the disease comprises thrombocytopenia, microangiopathic hemolytic anemia, and acute kidney injury. In most cases, aHUS is caused by genetic mutations in components of the alternative complement pathway. The risk of graft loss in patients after kidney transplantation with aHUS is dependent on the type of genetic mutation.
CASE REPORT: We present a case of a 32-year-old patient after a second kidney transplantation with atypical hemolytic uremic syndrome, whose clinical manifestation was triggered by the Shiga toxin-producing E. coli infection. Genetic testing revealed a new mutation (p.I342T) in the gene encoding complement factor B (CFB). Since 2 causative variants for aHUS have been described in the same exon of CFB gene, it might be supposed that the p.I342T variant has similar deleterious effects on CFB function. Despite the implemented treatment, graft function deteriorated and the patient had to return to a hemodialysis program and is currently on a waiting list for a third kidney transplant. The presence of the gene variant with increased susceptibility for aHUS and its recurrence after kidney transplantation makes the patient a good candidate for therapy with complement factor C5 inhibitors during and after the planned kidney transplantation.
CONCLUSIONS: Our case confirms the importance of genetic testing in patients with any sign of thrombotic microangiopathy. The finding of Shiga toxin-induced HUS with typical clinical course should not limit our vigilance of complement-mediated HUS with high risk of renal failure.
Keywords: Atypical Hemolytic Uremic Syndrome, Purpura, Thrombotic Thrombocytopenic, Shiga Toxin, eculizumab, Kidney Transplantation, Humans, Adult, Complement Factor B, Escherichia coli, Thrombotic Microangiopathies, Mutation, Escherichia coli Infections
Background
Hemolytic uremic syndrome (HUS) is typically characterized by thrombocytopenia, microangiopathic hemolytic anemia, and acute kidney injury. The clinical symptoms of HUS result from thrombotic microangiopathy (TMA), in which a thickening of the walls of small vessels (arterioles and capillaries), edema, and detachment of the endothelium are observed [1]. Thrombi lead to obstruction of the vessel lumen, damaging many tissues and organs, mainly the kidneys [1].
In clinical practice, the most commonly used classification is the division into typical, atypical, and secondary HUS [2]. The term ‘typical HUS’ is used for the distinctive form caused by Shiga toxin-induced endothelial cell damage produced by
In contrast, atypical hemolytic uremic syndrome (aHUS) is an ultra-rare disorder characterized by over-activation and dys-regulation of the alternative complement pathway [4]. Most cases of complement-related HUS arise from genetic mutations in complement factors, and only 5–6% are caused by antibodies against complement proteins [5–7]. Complement factor H (CFH), complement factor I (CFI), membrane cofactor protein (MCP or CD46), and thrombomodulin (THBD), which are regulatory proteins of the alternative complement pathway, and C3 and complement factor B (CFB), which are the C3 convertase proteins, are directly involved in the pathogenesis of the disease [2]. Complement factor B is cleaved by factor D into 2 fragments: Ba and Bb. Bb fragment, a serine protease, then combines with complement factor 3b to generate the C3 or C5 convertase. It has been implicated in proliferation and differentiation of preactivated B-lymphocytes, rapid spreading of peripheral blood monocytes, stimulation of lymphocyte blastogenesis, and lysis of erythrocytes. Ba inhibits the proliferation of preactivated B-lymphocytes [8].
Mutations in the genes encoding these proteins lead to uncontrolled activation of the alternative complement pathway, which cause excess production of the membrane attack complex (C5b-C9 complex) and C5, eventually leading to widespread endothelial injury and organ damage and failure [2,7,9]. CFB gene localizes to the major histocompatibility complex (MHC) class III region on chromosome 6. This cluster includes several genes involved in regulation of immune reaction [10].
Thrombotic microangiopathy is a rare but severe complication of kidney transplantation. De novo TMA affects 2.8–3.5% of kidney transplant recipients and is associated with a 22% probability of graft loss [11].
The risk of aHUS relapse following kidney transplantation varies according to the underlying genetic defects. Abnormalities in circulating complement system proteins, CFH, CFI, CFB, and C3 are associated with a high risk of disease recurrence because these proteins remain in the blood after transplantation [12,13]. The risk of relapse is much lower in patients with isolated abnormalities of membrane and intracellular cofactor proteins and diacylglycerol kinase because protein expression on the surface of the transplanted organ is normal [14,15].
HUS associated with Shiga toxin (STEC-HUS) does not recur after transplantation in adults, but in children there have been reports of its recurrence [16] since Shiga toxin-neutralizing antibodies remain in circulation for a long time. In contrast, the forms of HUS that are not related to Shiga toxin carry a significant risk of recurrence and graft loss after kidney transplantation in children and adults. Before the introduction of treatment with complement system inhibitors, the recurrence rate of atypical HUS after kidney transplantation was estimated at 60–80% [17–19]. In adults, aHUS recurrence occurs early in the post-transplant period, most often in the first 2 months after transplantation [17–19].
Genetic screening for complement mutations is indicated in patients with end-stage renal disease caused by aHUS who are eligible for kidney transplantation. Initiation of therapy with complement factor C5 inhibitor in the time and after kidney transplantation decreases the risk of aHUS recurrence in the graft.
Case Report
At the age of 8 years, the patient was diagnosed with kidney disease presenting with proteinuria and microscopic hematuria without impaired kidney function. Due to the abnormal urinalysis results, a kidney biopsy was performed, which revealed membranoproliferative glomerulonephritis. Despite immunosuppressive treatment, remission of the disease was not achieved and a gradual deterioration of renal function was observed, eventually leading to end-stage renal disease and the need to initiate renal replacement therapy. After 2 years of hemodialysis treatments, the patient received a kidney transplant from a deceased donor. Two months later, decreased diuresis, impaired graft function, and the development of massive protein-uria were observed. An increase of vascular flow resistance was seen in graft ultrasound; therefore, immunosuppressive treatment was intensified with the use of high-dose intravenous steroids combined with a series of plasmapheresis. The treatment resulted in an initial increase of diuresis and improvement of graft function, but graft function worsened 1 month later. At that time, the first graft biopsy was conducted, which revealed retrograde changes in tubular cells with oxalate deposits in the tubules, but without any signs of rejection. Soon thereafter, a Doppler ultrasound showed an absence of arterial blood flow in the kidney graft, with only minimal venous flow. Because of the fever that developed in the next few days, it was decided to perform exploratory laparotomy along with revision of the transplanted kidney. Macroscopic necrosis of the transplanted kidney was found and the graft was removed. Microscopic examination of the removed graft showed graft necrosis. In the lumen of the vessels, clots with partial recanalization were found. The patient had to restart dialysis.
Four years later, at the age of 14 years, the patient received a second kidney transplant. The kidney transplanted from the deceased donor resumed its function immediately after surgery and the postoperative period was uneventful. The patient received a triple immunosuppressive therapy consisting of prednisone, mycophenolate mofetil, and tacrolimus. The post-transplant course was complicated by several cytomegalovirus infections, all of which were successfully treated with intravenous ganciclovir. Six years later, a gradual deterioration of graft function was observed, accompanied by severe anemia, inadequate to the degree of graft dysfunction. A biopsy of the transplanted kidney was performed, in which electron microscopy revealed typical features of thrombotic microangiopathy. Calcineurin inhibitors were discontinued because they may have been a secondary cause of the disease. However, further deterioration of graft function was observed. After another 3 years, the patient was admitted to the hospital due to high blood pressure, decreased urine output, and generalized edema. Laboratory tests revealed severe anemia, with hemoglobin 6.9 g/dl and thrombocytopenia. In addition, features of hemolysis were observed in laboratory tests. Schistocytes were present in a blood smear together with greatly increased serum lactic dehydrogenase and decreased plasma haptoglobin. The stool samples were collected for Shiga toxin and blood samples for disintegrin and metal-loproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS-13) activity and for genetic testing. Another biopsy of the transplanted kidney was performed, which confirmed the diagnosis of chronic thrombotic microangiopathy (Figure 1). Plasma exchanges and infusions of fresh frozen plasma were started. Due to the persistent oliguria, increasing fluid overload and graft failure hemodialysis therapy had to be initiated. A fecal Shiga toxin test was routinely ordered and surprisingly tested positive. This prompted us to discontinue plasma exchange therapy. Further follow-up showed a stabilization of blood hemoglobin concentration at the level of 9–10 g/dL and platelet count at 90–100×103/µL. Schistocytes continued to be present in blood smears. Despite an increase of daily diuresis, graft function worsened, and the patient had to return to hemodialysis treatment. The case report timeline is provided in Figure 2.
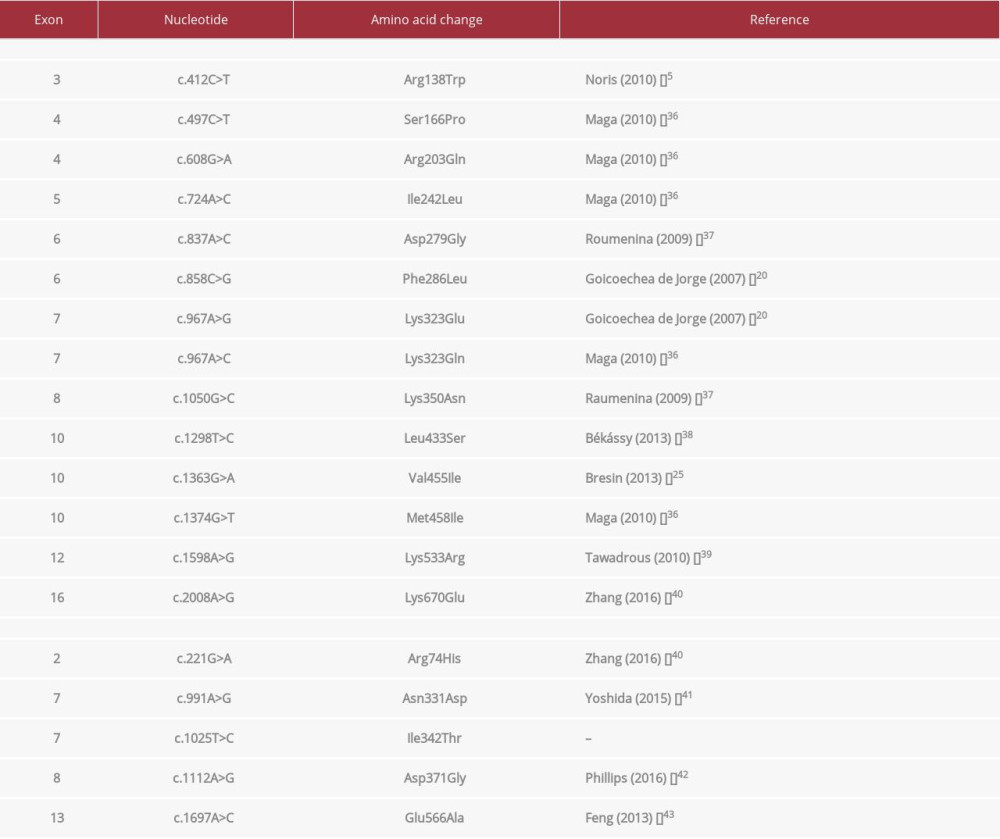
As part of genetic testing, DNA sequence of the whole coding regions of complement factors, including complement factor B gene (CFB, exons 1–18), was determined by direct DNA sequencing of polymerase chain reaction (PCR) products amplified from total genomic DNA. It identified previously unknown mutation in the gene encoding complement factor B (CFB) – the missense mutation p.I342T. Since 2 causative variants for aHUS have been described in exon 7 of CFB gene, it might be supposed that the p.I342T variant is likely causative for aHUS, having similar effects on CFB function, leading to decreased regulation by CFH and CFI. Causality of this mutation was checked using Polyphen-2 application. The mutation was defined as “possibly damaging” with score: 0.911 (sensitivity: 0.81; specificity: 0.94) by the HumDiv model. Currently known CFB gene point mutations identified as causative or likely causative of the atypical HUS phenotype are provided in Table 1, along with their schematic location in particular domains of CFB (Figure 3).
Discussion
In this report we describe a case of a patient who underwent 2 kidney transplants and was eventually diagnosed with aHUS caused by a novel mutation. The appearance of aHUS in the second kidney transplant was most likely triggered by the Shiga toxin-producing
The mutations in CFB are associated with a loss of kidney function in 88% of patients [17]. Although our patient had CFB mutations, aHUS was neither identified as a cause of end-stage renal disease in childhood nor as a cause of the first kidney transplant loss. No features of hemolysis were observed until
Our patient was also a carrier of several aHUS common risk variants (CFH promoter SNP, H3 haplotype, and C3 SNP). The CFB mutation and aHUS risk variants identified in our patient jointly represent an increased risk of the dysregulation of the alternative complement pathway causing aHUS [21]. Therefore, based on the clinical picture, histopathological changes, and the described genetic variants, the patient was diagnosed with aHUS.
In aHUS, the risk of disease recurrence is determined by a genetic mutation in the genes encoding the complement proteins [4,20,22–25]. Mutation-based risk stratification is very important for making decisions about post-transplant management in patients with aHUS. A greater risk of relapse is found in patients with relapse in previous transplants and in the carriers of pathogenic variants in CFH, CFB, CFH: CFHR1, TBHD rearrangements. There is moderate risk in carriers of CFI, C3 variants, anti-FH antibodies, homozygous for CFH-H3 haplotypes, no variants [24–31] and low risk in people with isolated MCP, DGKE variants, and negative anti-FH antibodies at the time of transplant [31–34].
To date, one of the most effective treatment regimens for aHUS is plasmatherapy consisting of plasma exchange and/or plasma infusion [21,35]. Our patient received plasma exchange as an empirical treatment for an acute rejection after the first transplant, and this treatment stabilized the graft function. However, after discontinuation of this therapy, the function of the transplanted kidney deteriorated again. The kidney biopsy neither showed a rejection nor a vascular microangiopathy; therefore, plasma exchange treatment was not indicated. The patient lost her first transplant very quickly, after 2 months. Imaging studies showed the disturbances in the vessels of the transplanted kidney, which already at that time could suggest vascular microangiopathy, which was, however, not confirmed in the biopsy. After the second transplant, the graft function has been stable for the first several years, but later started to gradually decline. After 14 years, a kidney biopsy was performed and confirmed the diagnosis of thrombotic microangiopathy. Due to suspected drug-related TMA, the immunosuppressive regimen was modified, but did not prevent further worsening of graft function. At that time, genetic tests were also performed and the diagnosis of aHUS was eventually established.
In our case it was too late to administer the C5 complement inhibitor, and the confirmation of the presence of CFB mutation occurred when the patient already required dialysis because of graft failure. The patient is currently waiting for a third kidney transplant and is scheduled to start eculizumab therapy in the peri-transplant period.
Our case report also describes a new heterozygous CFB variant that most likely predisposed our patient to the loss of a transplanted kidney due to TMA. This new variant is another that causes TMA with an increased risk of recurrence in the kidney transplant. The functional significance and pathogenicity of the reported change will require confirmation in other patients with aHUS.
Conclusions
Our case confirms the importance of genetic testing in patients with any signs of thrombotic microangiopathy. The finding of Shiga toxin-induced HUS with typical clinical course should not limit our vigilance of complement-mediated HUS with high risk of renal failure.
Figures
References:
1.. Rosove M, Thrombotic microangiopathies: Semin Arthritis Rheum, 2014; 43; 797-805
2.. Canpolat N, Hemolytic uremic syndrome: Turk Pediatri Ars, 2015; 50(2); 73-82
3.. George J, Nester C, Syndromes of thrombotic microangiopathy: N Engl J Med, 2014; 371; 654-66
4.. Barbour T, Johnson S, Cohney S, Hughes P, Thrombotic microangiopathy and associated renal disorders: Nephrol Dial Transplant, 2012; 27(7); 2673-85
5.. Noris M, Caprioli J, Bresin E, Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype: Clin J Am Soc Nephrol, 2010; 5; 1844-59
6.. Geerdink LM, Westra D, van Wijk JA, Atypical hemolytic uremic syndrome in children: Complement mutations and clinical characteristics: Pediatr Nephrol, 2012; 27(8); 1283-91
7.. Dragon-Durey MA, Loirat C, Cloarec S, Anti-factor H autoantibodies associated with atypical hemolytic uremic syndrome: J Am Soc Nephrol, 2005; 16(2); 555-63
8.. , UniProt Protein Summary for Protein P00751-CFAB_Human (Complement factor B (human)) [Internet] Available at: (accessed on 28-May-2022)https://beta.uniprot.org/uniprotkb/P00751/entry#function
9.. Loirat C, Frémeaux-Bacchi V, Atypical hemolytic uremic syndrome: Orphanet J Rare Dis, 2011; 6; 60
10.. , CFB complement factor B [Homo sapiens (human)] Gene ID: 629, updated on 5-Jun-2022 Available at: (accessed on 28-May-2022)https://www.ncbi.nlm.nih.gov/gene/629
11.. Salvadori M, Bertoni E, Update on hemolytic uremic syndrome: Diagnostic and therapeutic recommendations: Worl J Nephrol, 2013; 2(3); 56-76
12.. Schwimmer J, Nadasdy TA, Spitalnik PF, Kaplan KL, Xand MS, De novo thrombotic microangiopathy in renal transplant recipients: A comparison of hemolytic uremic syndrome with localized renal thrombotic microangiopathy: Am J Kidney Dis, 2003; 41(2); 471-79
13.. Noris M, Remmuzi G, Genetic abnormalities of complement regulators in hemolytic uremic syndrome: how do they affect patient management?: Nat Rev Nephrol, 2005; 1; 2-3
14.. Noris M, Remuzzi G, Hemolytic uremic syndrome: J Am Soc Nephrol, 2005; 16; 1035-50
15.. Fremeaux-Bacchi V, Blouin J, Complement factor I: A susceptibility gene for atypical haemolytic uraemic syndrome: J Med Gen, 2004; 41(6); 84
16.. Timothy HJ, Goodship M, Liszewski K, Mutations in CD46, a complement regulatory protein, predispose to atypical HUS: Trends Mol Med, 2004; 10; 226-31
17.. Loirat Ch, Niaudet P, The risk of recurrence of hemolytic uremic syndrome after renal transplantation in children: Pediatr Nephrol, 2003; 18(11); 1095-101
18.. Hebert D, Kim EM, Sibley RK, Mauer SM, Post-transplantation outcome of patients with hemolytic uremic syndrome: Update: Pediatr Nephrol, 1991; 5(1); 162-67
19.. Miller RB, Burke BA, Achmidt WJ, Recurrence of hemolytic uremic syndrome in renal transplants: A single center report: Nephrol Dial Transplant, 1997; 12(7); 1425-30
20.. Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome: Proc Natl Acad Sci USA, 2007; 104; 240-45
21.. Ahmed FA, El-Meanawy A, IgM nephropathy – successful treatment with rituximab: Saudi J Kidney Dis Transpl, 2019; 30; 235-38
22.. Noris M, Remuzzi G, Atypical hemolytic-uremic syndrome: N Engl J Med, 2009; 361; 1676-87
23.. Goodship TH, Cook HT, Fakhouri F, Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference: Kidney Int, 2017; 91; 539-51
24.. Bresin E, Daina E, Noris M, Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: Prognostic significance of genetic background: Clin J Am Soc Nephrol, 2006; 1; 88-99
25.. Bresin E, Rurali E, Caprioli J, European Working Party on Complement Genetics in Renal, Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype: J Am Soc Nephrol, 2013; 24; 475-86
26.. Le Quintrec M, Zuber J, Moulin B, Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome: Am J Transplant, 2013; 13; 663-75
27.. Loirat C, Fakhouri F, Ariceta G, An international consensus approach to the management of atypical hemolytic uremic syndrome in children: Pediatr Nephrol, 2016; 31; 15-39
28.. Le Quintrec M, Lionet A, Kamar N, Complement mutation-associated de novo thrombotic microangiopathy following kidney transplantation: Am J Transplant, 2008; 8; 1694-701
29.. Zuber J, Le Quintrec M, Morris H, Targeted strategies in the prevention and management of atypical HUS recurrence after kidney transplantation: Transplant Rev (Orlando), 2013; 27; 117-25
30.. Román-Ortiz E, Mendizabal Oteiza S, Pinto S, Eculizumab long-term therapy for pediatric renal transplant in aHUS with CFH/CFHR1 hybrid gene: Pediatr Nephrol, 2014; 29; 149-53
31.. Caroti L, Di Maria L, Carta P, Posttransplant outcome of atypical haemolytic uraemic syndrome in a patient with thrombomodulin mutation: A case without recurrence: Clin Kidney J, 2015; 8; 329-31
32.. Valoti E, Alberti M, Tortajada A, A novel atypical hemolytic uremic syndrome-associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H-dependent complement regulation: J Am Soc Nephrol, 2015; 26; 209-19
33.. Azukaitis K, Simkova E, Majid MA, The phenotypic spectrum of nephropathies associated with mutations in diacylglycerol kinase: J Am Soc Nephrol, 2017; 28; 3066-75
34.. Bresin E, Rurali E, Caprioli J, Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype: J Am Soc Nephrol, 2013; 24; 475-86
35.. Khandelwal P, Sinha A, Hari P, Outcomes of renal transplant in patients with anti-complement factor H antibody-associated hemolytic uremic syndrome: Pediatr Transplant, 2014; 18; 134-39
36.. Maga TK, Nishimura CJ, Weaver AE, Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome: Hum Mutat, 2010; 31; 1445-60
37.. Roumenina LT, Jablonski M, Hue C, Hyperfunctional C3 convertase leads to complement deposition on endothelial cells and contributes to atypical hemolytic uremic syndrome: Blood, 2009; 114; 2837-45
38.. Békássy ZD, Kristoffersson A, Cronqvist M, Eculizumab in an anephric patient with atypical haemolytic uraemic syndrome and advanced vascular lesions: Nephrol Dial Transplant, 2013; 28; 2899-907
39.. Tawadrous H, Maga T, Sharma J, A novel mutation in the complement factor B gene (CFB) and atypical hemolytic uremic syndrome: Pediatr Nephrol, 2010; 25; 947-51
40.. Zhang T, Lu J, Liang S, Comprehensive analysis of complement genes in patients with atypical hemolytic uremic syndrome: Am J Nephrol, 2016; 43; 160-69
41.. Yoshida Y, Miyata T, Matsumoto M, A novel quantitative hemolytic assay coupled with restriction fragment length polymorphisms analysis enabled early diagnosis of atypical hemolytic uremic syndrome and identified unique predisposing mutations in Japan: PLoS One, 2015; 10; e0124655
42.. Phillips EH, Westwood JP, Brocklebank V, The role of ADAMTS-13 activity and complement mutational analysis in differentiating acute thrombotic microangiopathies: J Thromb Haemost, 2016; 14; 175-85
43.. Feng S, Eyler SJ, Zhang Y, Partial ADAMTS13 deficiency in atypical hemolytic uremic syndrome: Blood, 2013; 122; 1487-93
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133