18 September 2022: Articles 
Combined Factor V and VIII Deficiency with LMAN1 Mutation: A Report of 3 Saudi Siblings
Challenging differential diagnosis, Rare disease
Shahad Alsheikh1AEF*, Rizam Alghamdi1AEF, Ahlam Alqatari12ABD, Abdullah Alfareed12ABD, Mona AlSaleh12ABDEDOI: 10.12659/AJCR.937312
Am J Case Rep 2022; 23:e937312






Abstract
BACKGROUND: Combined factor V and factor VIII deficiency (F5F8D) is a rare bleeding disorder with an incidence of 1: 1 000 000. The identified mutations were observed in LMAN1 and MCFD2 genes. This case report presents the cases of 3 Saudi siblings with the genetic mutation of LMAN1 causing F5F8D, and highlights the challenges in diagnosis and treatment.
CASE REPORT: Patient X, a 7-year-old boy, was misdiagnosed with hemophilia A after a history of prolonged circumcision bleeding and epistaxis. He was referred to our clinic for pre-operative assessment. Blood workup showed prolonged PT and aPTT, which were normalized by mixing studies. Since his previous diagnosis could not explain a prolonged PT, further investigations were performed, revealing low levels of FVIII and FV. Genetic testing confirmed a c.822G>A homozygous LMAN1 mutation. The other 2 siblings (patient Y and Z), who were 5- and 12-year-old, respectively, girls, were also assessed. They both had a history of epistaxis. The younger sibling also had an episode of bleeding after tooth extraction, and physical examination of this patient revealed a bruise over her left thigh. The older sibling had menorrhagia. Blood workup of both revealed prolonged PT and aPTT, with complete correction by mixing study, and low levels of FV and FVIII. The patients’ backgrounds and lab results were highly suggestive of F5F8D.
CONCLUSIONS: This case report describes an extremely rare bleeding disorder. More attention should be directed toward this disease, and a careful evaluation of suspicious cases should be performed to better diagnose and manage these patients.
Keywords: Familial Multiple Coagulation Factor Deficiency I, LMAN1 Protein, Human, Child, Child, Preschool, Epistaxis, Factor V, Factor V Deficiency, Female, Hemophilia A, Humans, Male, Mannose-Binding Lectins, Membrane Proteins, Mutation, Saudi Arabia, Siblings, Vesicular Transport Proteins
Background
Combined factor V and factor VIII deficiency (F5F8D) is a rare genetic bleeding disorder that accounts for 3% of all congenital bleeding conditions, with an incidence of 1: 1 000 000 [1–3]. F5F8D is an autosomal recessive disease, while the isolated deficiency of factor V and factor VIII is autosomal recessive and X-linked, respectively [1,2]. The identified mutations were observed in Lectin Mannose Binding Protein (LMAN1) and Multiple Coagulation Factor Deficiency 2 (MCFD2) genes [2–4]. While the LMAN1 gene is located in chromosome 18q21, and MCFD2 is located in chromosome 2p21, they both share the same functional pathway carrying both factors V and VIII from the endoplasmic reticulum (ER) to the Golgi apparatus. Therefore, mutation in either protein will result in a transportation defect, hence retaining the coagulation factors within the cell and causing ineffective thrombogenesis [2,3,5,6].
The disease is more prevalent in Middle-Eastern, Mediterranean, and South Asian countries, as the most notable risk factor is consanguineous marriage [1–3]. Whereas LMAN1 mutation is more common in the Middle East, MCFD2 is more common in India and Europe [1,2]. In a comprehensive review of F5F8D, most reported cases had mild-to-moderate bleeding symptoms. About 60–80% of F5F8D patients are discovered by incidental findings after surgery or by prolonged trauma bleeding. The clinical symptoms can overlap with other bleeding disorders. However, the most commonly associated symptoms with F5F8D are menorrhagia, epistaxis, and extensive bleeding secondary to surgical procedures or trauma, and to a lesser extent bleeding into joints, the gastrointestinal tract, and intracranially [2,3].
The normal range of FV and FVIII is above 30–40%, and a marked decrease in the serum levels of these factors, commonly noted in F5F8D patients to be less than 30%, is a diagnostic clue [1–3,5]. Nevertheless, in a deficient subject, the level of the factor can be as low as 1% [7]. Another diagnostic tool is the prolonged indices, which are the prothrombin time (PT) and activated partial thromboplastin time (aPTT) [2,3,7,8]. Management aims to restore factors based on the severity of the bleeding episode or as prophylaxis, which can be achieved by fresh frozen plasma (FFP), desmopressin (DDAVP), and plasma-derived or recombinant factors. Each management option can be used differently; FFP can restore both factors, but it is commonly used to optimize factor V levels. Various methods can replenish FVIII, yet the most common is desmopressin. It has been documented that the FVIII level increases by more than 2-fold within 1–2 h after administering DDAVP in a F5F8D patient [1–3]. Our case report aims to shed light on the challenges in the diagnosis and management of F5F8D in 3 Saudi siblings.
Case Reports
PATIENT X:
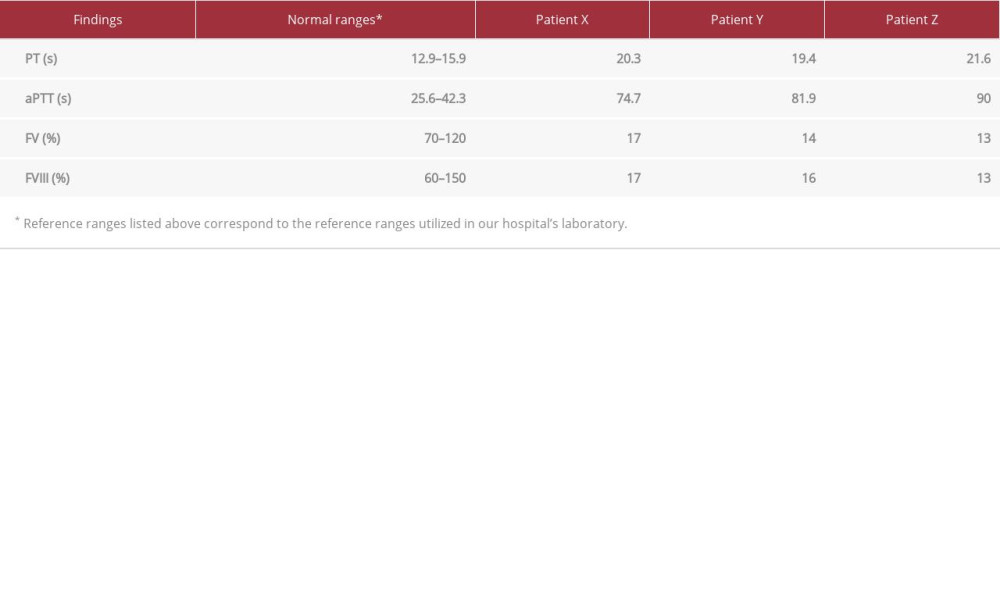
Our patient, referred to as patient X for the patient’s confidentiality, was a 7-year-old Saudi boy who was misdiagnosed with hemophilia A after a history of prolonged circumcision bleeding. He was referred to our clinic for pre-operative assessment for eustachian tube dysfunction. Further history-taking revealed epistaxis. The physical examination was unremarkable. Blood workup (Table 1) showed prolonged PT and aPTT, which were normalized by mixing studies. Since a prolonged PT could not be explained by his previous diagnosis, we ordered more investigations, and low levels of FVIII and FV were reported. The abnormally low FV and FVIII levels suggested a need for genetic testing. The genetic test requested for this patient was the coagulation deficiency NGS panel, which is used to diagnose inherited bleeding disorders. The test confirmed a homozygous c.822G>A mutation in the LMAN1 gene, previously identified in several Iranian patients. This mutation did not change the amino acid corresponding to the modified codon, but caused a splicing defect between exon 7 and exon 8 [9]. The patient’s parents, who are consanguineous, and his only 2 siblings, were further investigated (Figure 1). The siblings are referred to here as patient Y and patient Z.
PATIENT Y:
Although the parent’s results revealed normal PT, aPTT, mixing studies, and factor levels, the siblings’ results were abnormal. Patient Y, a younger sibling of patient X, was a 5-year-old Saudi girl. She had a history of an episode of bleeding after tooth extraction and recurrent history of epistaxis. In addition, physical examination revealed a bruise on her left thigh. Further laboratory testing (Table 1) confirmed prolonged PT and aPTT, with complete correction by mixing study, and low levels of FV and FVIII.
PATIENT Z:
Patient Z, a 12-year-old Saudi girl, was an elder sibling of patient X. She had a history of epistaxis and menorrhagia. Upon laboratory evaluation (Table 1), similarly, the patient had prolonged PT and aPPT, which were corrected by mixing studies. FV and FVIII levels were markedly low.
All 3 patients’ investigations included complete blood count, platelet function assay, ristocetin cofactor assay, von Willebrand factor antigen assay, thrombin time, and factor X and VII as-says to aid in the differential diagnosis. However, they all revealed unremarkable results. In addition, neither the parents or the other siblings underwent genetic testing. The parents refused to take the genetic test, and the siblings were diagnosed based on the clinical and laboratory findings.
Our 3 patients received case-based treatment. The bleeding episodes in all 3 were not severe, and no further treatment was required. Preventive measures, such as factors replacement, were recommended before any surgical intervention. However, the eldest sister required oral tranexamic acid to manage her menorrhagia.
Discussion
F5F8D is a rare autosomal recessive bleeding disorder, accounting for approximately 3% of congenital bleeding disorders. The disease was first identified in 1954 by Oeri et al [7]. Forty-seven identified mutations were noted in both LMAN1 and MCFD2 genes, accounting for 68% and 32%, respectively. Though the mutations were found on 2 different genes, the phenotype was noted to be highly similar [1,6]. However, a study conducted in 2008 showed that levels of factor FV and factor FVIII were lower in patients with identified mutations in MCFD2 gene than LMAN1 gene. Thus, more studies are required to establish the correlation between genotype-pheno-type in F5F8D patients [10].
Bleeding in F5F8D was reported in the literature to be mild-to-moderate in nature, with the most common reported symptoms being menorrhagia, epistaxis, and extensive bleeding secondary to surgical procedures or trauma [2,3,11]. Similarly, we have observed that our patients presented with mild bleeding episodes; in fact, 2 of them were only diagnosed after screening. Based on this observation, we may conclude that unlike what had been observed in patients with hemophilia A in clinical practice, patients with F5F8D have a less severe disease; however, this needs to be validated in the future by studies that compare F5F8D and other congenital factor deficiencies. Furthermore, as of now, there is a gap in the literature regarding many aspects of this disease due to its rarity, and more studies are required to bridge that gap.
Generally, bleeding signs and symptoms are common; thus, physicians tend to prioritize common differential diagnoses. However, although a patient may have a common problem, the underlying disease may be extremely rare, and, as seen in our cases, the signs and symptoms of this rare disease are unspecific and can be shared with other common and rare bleeding disorders. Therefore, reaching an accurate diagnosis can be challenging. As a result, some patients live underdiagnosed or with an inaccurate diagnosis, as seen in our 3 cases. Nevertheless, reaching an accurate diagnosis is essential, especially in genetic bleeding disorders, as it will aid in providing genetic counseling and screening for the whole family, and an appropriate treatment will be provided for each patient. These steps will enhance patient care and potentially reduce complication rates.
A standardized treatment and prophylactic measures for this disease have not been developed due to the rarity of the disease and the scarcity of data. The mild-to-moderate nature of the disease does not indicate regular treatment. The management aims to restore factors based on the severity of the bleeding episode or as prophylaxis, which can be achieved by FFP, DDAVP, plasma-derived, or recombinant factors [2,3].
FFP can restore both factors, but it is commonly used to optimize factor V levels. Nevertheless, the use of FFP have been associated with volume overload [12]. Moreover, regarding factor V, a recent Chinese study found that low level of factor V is associated with low levels of the anticoagulant protein tissue factor pathway inhibitor (TFPI) [13]. As a result, low levels of factor V in F5F8D can balance the bleeding tendency that occurs due to FVIII deficiency. Various methods can replenish FVIII, yet the most common is desmopressin, with peak efficacy within half an hour. It has been documented that the FVIII levels increase by more than 2-fold within 1–2 h by administering DDAVP in F5F8D patients [2,3,7,14]. In addition, this Chinese study has shown a complete response in 5 out of 6 patients with the use of DDAVP only [13].
Replenishing the factors levels can be achieved using various modalities. However, the recent Chinese study suggested the use of DDAVP to restore FVIII without the need for FFP [13]. More studies are required to evaluate the efficacy of each treatment modality and to develop guidelines to provide better and efficient care of the patients.
Prophylactic measures are still under development; however, some studies have suggested prenatal screening for carrier parents or the ones with severely affected offspring. Genetic testing could be achieved by DNA extraction from the chorionic villi at 10–12 gestational weeks. However, it is crucial to mention that the disease’s nature is not severe; hence, screening for the general population is not necessarily recommended without strong risk factors [1]. Another prophylactic measure is factor replenishment prior to surgery, labor, or dental extractions. Prophylactic measures aim to reduce the risk of severe bleeding and complications. A review of 19 pregnant women with F5F8D, in which full findings were documented in 9 pregnancies, showed that postpartum hemorrhage occurred in 6 patients (32%). Two of these patients required blood transfusion, with 1 requiring 7 units, thus, shedding light on the role of prophylaxis in known cases of F5F8D [1,8].
Conclusions
F5F8D is an extremely rare bleeding disorder. More attention should be directed toward this disease, and a careful evaluation of suspicious cases should be performed to better diagnose and manage these patients. A limitation of this report is that genetic testing for LMAN1 was not performed for the parents or siblings.
References:
1.. Spreafico M, peyvandi F, Combined FV and FVIII deficiency: Haemophilia, 2008; 14(6); 1201-8
2.. Chapin J, Cardi D, Gibb C, Laurence J, Combined factor V and factor VIII deficiency: A report of a case, genetic analysis, and response to desmopressin acetate: Clin Adv Hematol Oncol, 2012; 10(7); 472-74
3.. Zheng C, Zhang B, Combined deficiency of coagulation factors V and VIII: An update: Semin Thromb Hemost, 2013; 39(06); 613-20
4.. Zhang B, Combined deficiency of factor V and factor VIII is due to mutations in either LMAN1 or MCFD2: Blood, 2006; 107(5); 1903-7
5.. Nichols W, Seligsohn U, Zivelin A, Mutations in the ER-Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII: Cell, 1998; 93(1); 61-70
6.. Zhang B, Cunningham M, Nichols W, Bleeding due to disruption of a cargo-specific ER-to-Golgi transport complex: Nat Genet, 2003; 34(2); 220-25
7.. Spiliopoulos D, Kadir R, Congenital factor V and VIII deficiency in women: Blood Coagul Fibrinolysis, 2016; 27(3); 237-41
8.. Viswabandya A, Baidya S, Nair S, Clinical manifestations of combined factor V and VIII deficiency: A series of 37 cases from a single center in India: Am J Hematol, 2010; 85(7); 538-39
9.. Neerman-Arbez M, Johnson K, Morris M, Molecular analysis of the ERGIC-53 gene in 35 families with combined factor V-factor VIII deficiency: Blood, 1999; 93(7); 2253-60
10.. Zhang B, Spreafico M, Zheng C, Genotype-phenotype correlation in combined deficiency of factor V and factor VIII: Blood, 2008; 111(12); 5592-600
11.. Peyvandi F, Tuddenham E, Akhtari A, Bleeding symptoms in 27 Iranian patients with the combined deficiency of factor V and factor VIII: Br J Haematol, 1998; 100(4); 773-76
12.. Mannucci P, Duga S, Peyvandi F, Recessively inherited coagulation disorders: Blood, 2004; 104(5); 1243-52
13.. Shao Y, Wu W, Xu G, Wang X, Ding Q, Low factor V level ameliorates bleeding diathesis in patients with combined deficiency of factor V and factor VIII: Blood, 2019; 134(20); 1745-54
14.. Mansouritorghabeh H, Shirdel A, Desmopressin acetate as a haemostatic elevator in individuals with combined deficiency of factors V and VIII: A clinical trial: J Thromb Haemost, 2016; 14(2); 336-39
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133