15 November 2022: Articles 
Late-Onset Ornithine Transcarbamylase Deficiency Complicated with Extremely High Serum Ammonia Level: Prompt Induction of Hemodialysis as the Key to Successful Treatment
Challenging differential diagnosis, Management of emergency care, Rare disease
Satsuki Yamamoto12AEF, Shun YamashitaDOI: 10.12659/AJCR.937658
Am J Case Rep 2022; 23:e937658






Abstract
BACKGROUND: Ornithine transcarbamylase deficiency (OTCD) is an X-linked semi-dominant disorder, causing possible fatal hyperammonemia. Late-onset OTCD can develop at any time from 2 months after birth to adulthood, accounting for 70% of all OTCDs.
CASE REPORT: A 35-year-old man with chronic headaches stated that since childhood he felt sick after eating meat. Fourteen days before hospital admission, he began receiving 60 mg/day of intravenous prednisolone for sudden deafness. The prednisolone was stopped 5 days before hospital admission. Four days later, he was transferred to our hospital because of confusion. On admission, he had hyperammonemia of 393 µmol/L. Because he became comatose 7 hours after admission, and his serum ammonia increased to 1071 µmol/L, we promptly started hemodialysis. Because his family history included 2 deceased infant boys, we suspected late-onset OTCD. On day 2 of hospitalization, we began administering ammonia-scavenging medications. Because he gradually regained consciousness, we stopped his hemodialysis on day 6. After his general condition improved, he was transferred to the previous hospital for rehabilitation on day 32. We definitively diagnosed him with late-onset OTCD due to the low plasma citrulline and high urinary orotic acid levels found during his hospitalization.
CONCLUSIONS: Clinicians should suspect urea cycle disorders, such as OTCD, when adult patients present with marked hyperammonemia without liver cirrhosis. Adult patients with marked hyperammonemia should immediately undergo hemodialysis to remove ammonia, regardless of causative diseases.
Keywords: hyperammonemia, Renal Dialysis, Urea Cycle Disorders, Inborn, Male, Infant, Adult, Humans, Child, Ornithine Carbamoyltransferase Deficiency Disease, Ammonia, Prednisolone, Ornithine Carbamoyltransferase
Background
Ornithine transcarbamylase (OTC) deficiency (OTCD) is an X-linked semi-dominant disorder caused by mutations in the OTC gene on chromosome Xp21 [1]. OTCD is the most common urea cycle disorder (UCD), occurring in 1 in 56 500 people worldwide and 1 in 14 000 people in Japan [2,3]. Hyperammonemia resulting from OTCD-induced disturbances in the ammonia metabolism requires prompt treatment because it can cause disturbances of consciousness in severe cases despite usually showing only nonspecific and mild symptoms such as headache or appetite loss [4]. Among patients with OTCD, 30% are neonatal-onset type, and 70% are late-onset type [4]. Patients with neonatal-onset OTCD are mainly boys and can be detected by their manifestations of lethargy, vomiting, and seizures that worsen with their growth [5]. Without prompt and adequate treatment, the disease is usually fatal because of an almost complete deficiency of OTC [5]. Late-onset OTCD develops in men with partial OTC deficiency or heterozygous women at any time from 2 months after birth to adulthood when under stressors such as acute infections, menstruation, steroid administration, starvation, surgery, or increased protein intake [4–6]. To the best our knowledge, the median and oldest published ages of onset of late-onset OTCD were 35 and 69 years, respectively [3,7]. Late-onset OTCD is difficult to diagnose when patients show no signs or symptoms, because the clinical manifestations are nonspecific with normal laboratory findings except for marked hyperammonemia during attacks [2,4]. In these cases, unusual dietary habits such as avoiding meat consumption may help with the diagnosis.
Herein, we report a patient with late-onset OTCD with marked hyperammonemia, who we rescued by immediately starting hemodialysis, to emphasize the importance of suspecting OTCD when diagnosing hyperammonemia in adults without cirrhosis and prompt initiation of hemodialysis regardless of the causes of extreme hyperammonemia.
Case Report
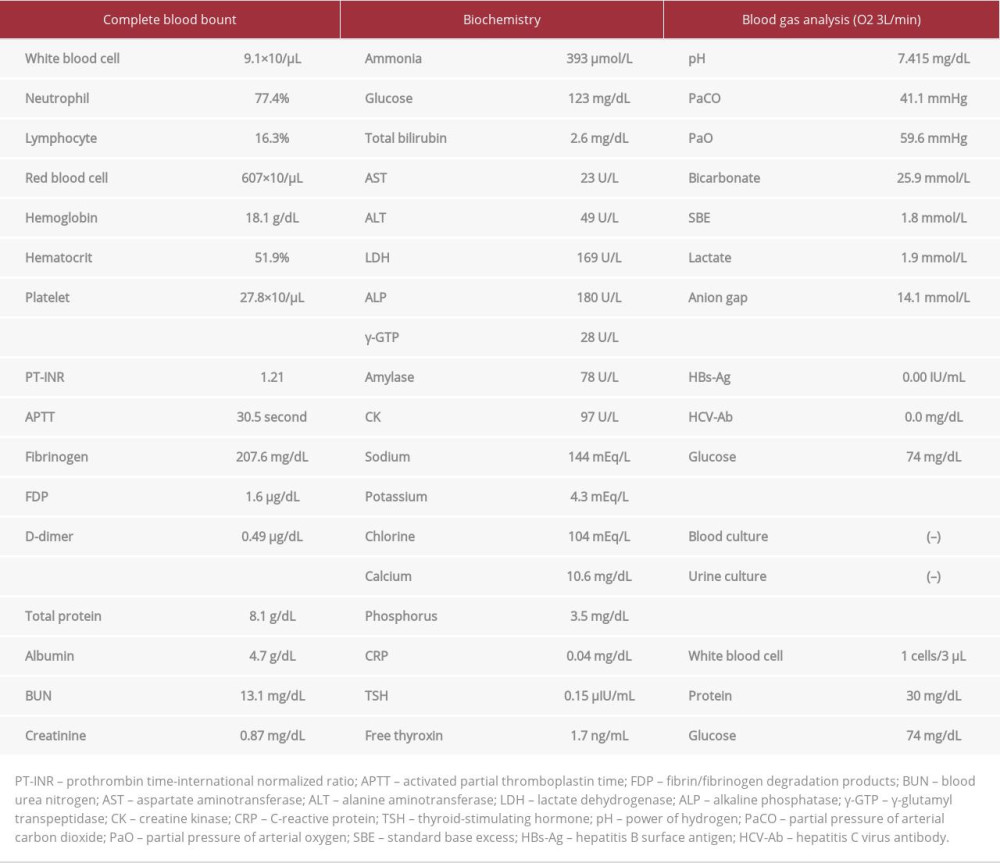
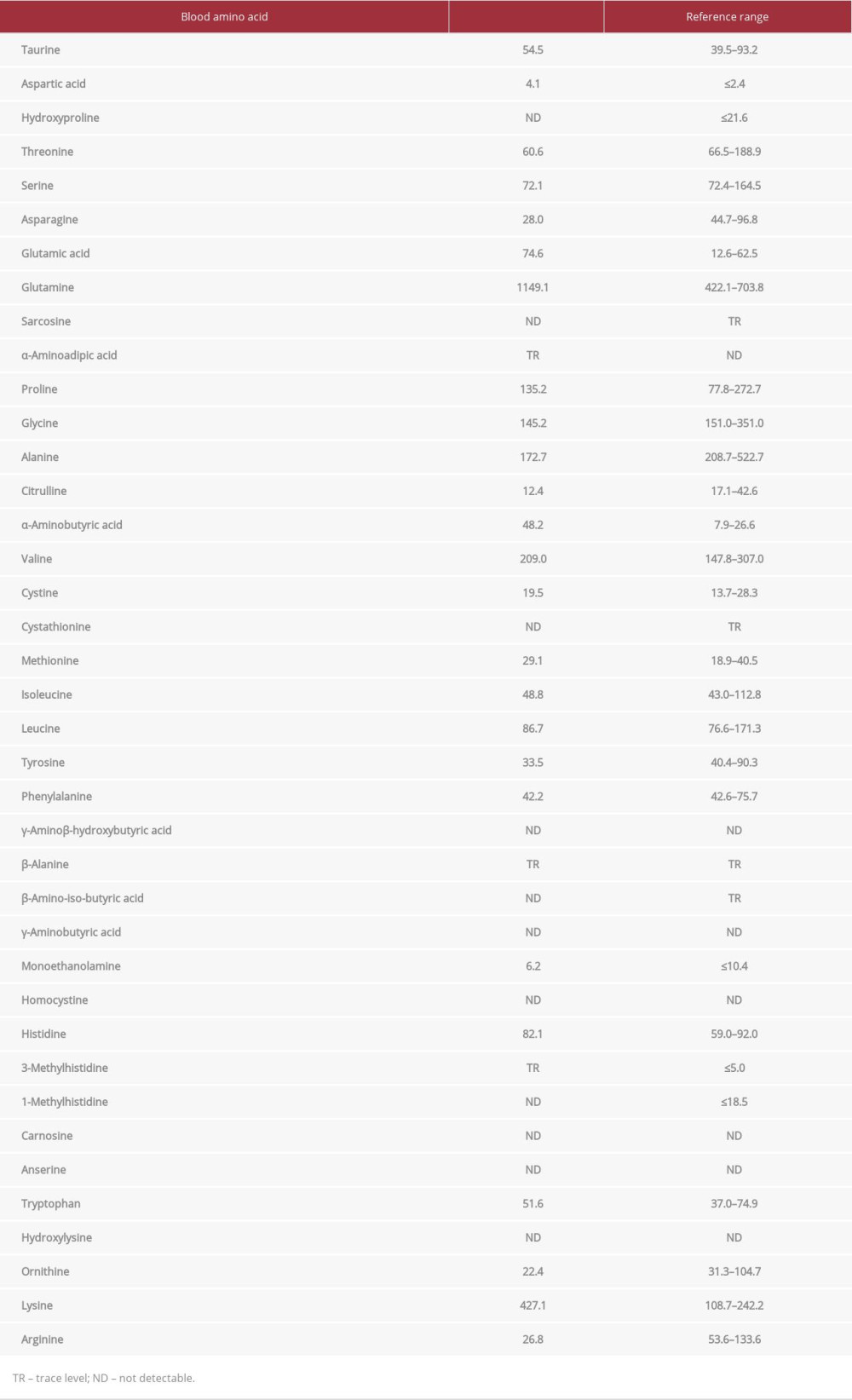
A 35-year-old man had been suffering from unexplained chronic headaches since childhood. He routinely avoided pork, beef, and other protein-rich foods because he felt sick after consuming them. Fourteen days prior to hospital admission, he had hearing loss in his right ear and visited a local otolaryngologist. He was diagnosed with sudden hearing loss and subsequently received intravenous administration of 60 mg/day predniso-lone. Although the prednisolone was tapered off and stopped 5 days prior to admission, he developed vomiting and a headache the next day. Four days later, he became confused and was transferred to our hospital. On admission, his vitals were a Glasgow Coma Scale (GCS) of 12 (E; Eye opening 3, V; Verbal response 4, M; Motor response 5), body temperature 36.9°C, pulse rate 100 beats/min, blood pressure 170/100 mmHg, respiratory rate 21/min, and oxygen saturation 98% with 3 L/min of oxygen via a nasal cannula. Physical examinations revealed no conjunctival icterus, with normal respiratory and cardiac sounds. His abdomen was flat and soft without hepatosplenomegaly. Only bilateral Chaddock reflexes were positive, without seizures or paralysis. Laboratory findings (Table 1) showed a white blood cell count of 9.1×103/µL, platelet count of 27.8×104/µL, C-reactive protein of 0.04 mg/mL, serum albumin level of 4.7 g/dL, serum creatine level of 0.87 mg/dL, aspartate aminotransferase level of 23 U/L, alanine amino-transferase level of 49 U/L, and fasting serum glucose level of 123 mg/dL. His initial serum ammonia level was markedly elevated to 393 µmol/L, which was considered to be the cause of his disturbance of consciousness. Arterial blood gas analysis with administration of 3 L/min of oxygen via a nasal cannula revealed a pH of 7.415, HCO3– of 25.9 mmol/L, and an anion gap of 14.1 mmol/L. No bacteria were detected via urinary culture. Spinal tap examination revealed an opening pressure of 21 cmH2O, with the cerebrospinal fluid being clear and colorless, with a white blood cell count of 1/3 µL. Cranial magnetic resonance imaging showed no abnormalities (Figure 1). Thoracic and abdominal computed tomography (CT) with contrast enhancement revealed no malignancies and a normal liver with no portosystemic shunt (Figure 2). UCD was suspected as the cause of the hyperammonemia at this time for the following reasons: extreme hyperammonemia with a normal anion gap, no hypoglycemia, no hepatic impairment, and family history of 2 infant boys who died of unknown causes among his mother’s siblings (2 men and 3 women). Seven hours after admission, his level of consciousness further deteriorated to GCS 3 (E1V1M1) with an extreme increase in his serum ammonia level to 1071 µmol/L. Hemodialysis was introduced 12 hours after admission because of an urgent need to decrease his serum ammonia level. Although the serum ammonia level improved to 523 µmol/L 3 hours after starting hemodialysis, he experienced generalized tonic convulsions. We concurrently started mechanical ventilation because profound sedation was required to stop the convulsions. We also administered 355 g/day glucose to prevent further elevating his serum ammonia levels owing to hypercatabolism. Beginning on day 2 of hospitalization, we prescribed 0.38 g/kg/day L-arginine to stimulate conversion of ammonia to urea and 9.4 g/m2/day sodium phenylbutyrate and 0.24 g/kg/day sodium benzoate as ammonia-scavenging therapy. On hospital day 3, despite his serum ammonia level decreasing to 68 µmol/L, cranial CT (Figure 3A, 3B) revealed whole-brain edema with an indistinct cortico-medullary junction at the base of the brain, which subsequently improved on cranial CT on day 5 (Figure 3C, 3D). We discontinued hemodialysis on day 6. On day 7, we received laboratory results concerning his urea cycle, which we had previously sent to an outside laboratory for examination. His urinary orotic acid and uracil levels as measured by gas chromatography/mass spectrometry were significantly elevated to 241.1 mmol/mol creatinine (reference range: 0.4–1.2) and 138.3 mmol/mol creatinine (reference range: 0–13), respectively. Blood amino acid analysis as performed by high performance liquid chromatography/mass spectrometry showed a high glutamine level of 1149.1 nmol/mL (reference range: 422.1–703.8), high glutamate level of 74.6 nmol/mL (reference range: 12.6–62.5), low citrulline level of 12.4 nmol/mL (reference range: 17.1–42.6), and low arginine level of 26.8 nmol/mL (reference range: 53.6–133.6); these findings are compatible with a diagnosis of delayed OTCD (Table 2). Additionally, genetic analysis of peripheral blood for hyperammonemia revealed a heterozygous pathogenic variant in Exon 2 of the
Discussion
OTCD is an X-linked semi-dominant disorder that causes hyperammonemia due to disturbances in ammonia metabolism [4]. Late-onset OTCD developing later than 2 months after birth is more common (approximately 70% of cases) than is infantile-type OTCD [4], making it an important differential diagnosis for adult patients with hyperammonemia. To make a correct diagnosis, clinicians must consider the following: history of unexplained chronic headache or hepatitis, characteristic dietary habits such as vegetarianism or avoiding protein intake, and unexplained deaths of third-degree or closer relatives, especially children [2,4]. However, obtaining such detailed medical or family histories can be difficult unless UCDs, including OTCD, are strongly suspected. We failed to gather information for our patient early after admission regarding his characteristic history, such as his unexplained chronic headaches since childhood, dietary habit of avoiding protein-rich foods, and unexplained deaths among his mother’s brothers. Although obtaining meticulous medical and family histories is important, it can be impractical to do so for all patients. Therefore, presence of other indicators to suggest late-onset OTCD would be of extreme value.
Marked hyperammonemia in adults may indicate late-onset OTCD. In adults, 90% of hyperammonemia is estimated to be caused by liver cirrhosis [11]. Possible causes of hyperammonemia in adults without liver cirrhosis include UCDs such as OTCD, portosystemic shunts, status epilepticus, malignancy, or infection with urease-producing bacteria [11,12]. Among these, OTCD can cause extreme marked hyperammonemia [4,13]. One study found that the median serum ammonia level of patients with late-onset OTCD at their initial visit was 240 µmol/L, while that of patients with acute liver failure with severe hepatic encephalopathy (grade III or higher) was 113 µmol/L [14]. When manifestations of OTCD develop after steroid administration, serum ammonia levels can be particularly high, the reported mean level being 1066 µmol/L [15]. Thus, late-onset OTCD exhibits much higher ammonia levels than those of acute liver failure with severe hepatic encephalopathy [4]. In addition, urinary uracil can increase in male patients with OTCD, making it a valid marker in such patients [16]. Our patient’s serum ammonia level on admission was extremely high (393 µmol/L), with a significantly high urinary uracil level and only mild liver damage. Therefore, marked hyperammonemia in adults could be a valuable indicator of a possible diagnosis of OTCD, especially in patients without liver cirrhosis. In such patients, it is also important to perform blood amino acid analysis and measure urinary organic acids, including urinary uracil, or perform genetic analysis for UCDs, including OTCD, in a timely manner.
Hyperammonemia can cause status epilepticus and comas, which leads to permanent higher cerebral dysfunction, especially in prolonged cases [2]. Therefore, clinicians must attempt to rapidly decrease serum ammonia levels to <150 µmol/L [2]. Survival rates of patients with peak serum ammonia levels >1000 µmol/L are lower than those of patients with levels <500 µmol/L. Hemodialysis is the first choice of treatment for such marked hyperammonemia [2,5]. Although the serum ammonia level indicated for introducing hemodialysis in adults is not clearly defined, hemodialysis should be introduced promptly when serum ammonia levels are increasing rapidly or exceed 350–400 µmol/L in neonatal UCDs [4]. In the present case, despite the rapid increase in the serum ammonia level from 393 µmol/L on admission to 1071 µmol/L, the patient was salvaged by prompt introduction of hemodialysis before a definite diagnosis of OTCD had been established.
Conclusions
Late-onset OTCD can be fatal when marked hyperammonemia is complicated. This can be induced by infections, steroid administration, or excessive protein intake. For adult patients, marked hyperammonemia without liver cirrhosis may be a valuable indicator of late-onset OTCD. Prompt introduction of hemodialysis is mandatory when patients present with extreme hyperammonemia, regardless of the cause.
Figures
References:
1.. Choi JH, Lee BH, Kim JH, Clinical outcomes and the mutation spectrum of the OTC gene in patients with ornithine transcarbamylase deficiency: J Hum Genet, 2015; 60(9); 501-7
2.. Nicholas AM, Kara LS, Andrea LG, Urea cycle disorders overview, 2003 (Last update: June 22, 2017). [Accessed March 2022]https://www.ncbi.nlm.nih.gov/books/NBK1217/
3.. Kana D, Takashi N, Yuko N, Late-onset ornithine transcarbamylase deficiency associated with hyperammonemia: Clin J Gastroenterol, 2017; 10(4); 383-87
4.. Anais B, Stephanie G, Jean BA, Long-term outcomes in ornithine transcarbamylase deficiency: A series of 90 patients: Orphanet J Rare Dis, 2015; 10; 58
5.. Barkovich E, Gropman AL, Late onset ornithine transcarbamylase deficiency triggered by an acute increase in protein intake: A review of 10 cases reported in the literature: Case Rep Genet, 2020; 2020; 7024735
6.. Cavicchi C, Donati M, Parini R, Sudden unexpected fatal encephalopathy in adults with OTC gene mutations-Clues for early diagnosis and timely treatment: Orphanet J Rare Dis, 2014; 9; 105
7.. Pizzi MA, Alejos D, Hasan TF, Adult presentation of ornithine transcarbamylase deficiency: Two Illustrative cases of phenotypic variability and literature review: Neurohospitalist, 2019; 9(1); 30-36
8.. Tuchman M, Plante RJ, McCann MT, Qureshi AA, Seven new mutations in the human ornithine transcarbamylase gene: Hum Mutat, 1994; 4(1); 57-60
9.. Cavicchi C, Donati M, Parini R, Sudden unexpected fatal encephalopathy in adults with OTC gene mutations-Clues for early diagnosis and timely treatment: Orphanet J Rare Dis, 2014; 9; 105
10.. Pinner JR, Freckmann ML, Kirk EP, Yoshino M, Female heterozygotes for the hypomorphic R40H mutation can have ornithine transcarbamylase deficiency and present in early adolescence: A case report and review of the literature: J Med Case Rep, 2010; 4; 361
11.. Upadhyay R, Bleck TP, Busl KM, Hyperammonemia: What urea-lly need to know: Case report of severe noncirrhotic hyperammonemic encephalopathy and review of the literature: Case Rep Med, 2016; 2016; 8512721
12.. Sakusic A, Sabov M, McCambridge AJ, Features of adult hyperammonemia not due to liver failure in the ICU: Crit Care Med, 2018; 46(9); e897-e903
13.. Rush ET, Hartmann JE, Skrabal JC, Rizzo WB, Late-onset ornithine transcarbamylase deficiency: An under recognized cause of metabolic encephalopathy: SAGE Open Med Case Rep, 2014; 2; 2050313 X14546348
14.. Bernal W, Hall C, Karvellas CJ, Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure: Hepatology, 2007; 46(6); 1844-52
15.. Imoto K, Tanaka M, Goya T, Corticosteroid suppresses urea-cycle-related gene expressions in ornithine transcarbamylase deficiency: BMC Gastroenterol, 2022; 22; 144
16.. Alsharhan H, Alharbi H, Priestley J, Urinary uracil: A useful marker for ornithine transcarbamylase deficiency in affected males: Clin Chem, 2020; 66(7); 988-89
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952791
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Most Viewed Current Articles
07 Dec 2021 : Case report
22,759,422
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  175,936
175,936
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,499
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,510
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133