04 November 2022: Articles 
Acute Intermittent Porphyria: Complete Phenotype in a Patient with p.Arg173Trp Variant in Thailand
Rare disease
Vachiravit Sriprakoon1BCDEF, Chalisa Ittagornpunth1BEF, Nakorn Puapaiboon1C, Aekasit Bunyahathaipat1C, Punnapat Piriyanon2BE, Sookkasem Khositseth2D, Kitiwan Rojnueangnit2ABCDEF*DOI: 10.12659/AJCR.937695
Am J Case Rep 2022; 23:e937695






Abstract
BACKGROUND: Acute intermittent porphyria (AIP) is a rare genetic disease caused by the deficiency of porphobilinogen deaminase enzyme in the heme synthesis pathway. AIP is passed by autosomal dominant inheritance. Heterozygous pathogenic variants in hydroxymethylbilane synthase (HMBS) are associated with AIP. Multisystemic manifestations of acute neurovisceral features exist, which are quite challenging for diagnosis. Currently, few patients worldwide have been reported with AIP. A small number of reports have been published in Thailand, but none have been confirmed by molecular genetics diagnosis.
CASE REPORT: A 14-year-old female adolescent presented with severe intermittent abdominal pain, vomiting, seizure, posterior reversible encephalopathy syndrome, syndrome of inappropriate antidiuretic hormone, and muscle weakness, which are all classic phenotypes of an acute AIP attack. The patient received several investigations before AIP was suspected. High levels of urine porphobilinogen, high levels of urine aminolevulinic acid, and a heterozygous known pathogenic variant in HMBS: c.517C>T (p.Arg173Trp) were identified. Therefore, AIP was the definitive diagnosis. Then, Sanger sequencing testing was performed for the patient’s family; this variant was found in her father, paternal grandmother, and sister, who were all asymptomatic (latent AIP). After the AIP was confirmed, high carbohydrate loading was given as a standard treatment. She had a full recovery; her clinical course of the attack episode lasted for 8 weeks.
CONCLUSIONS: An early diagnosis of AIP leads to prompt and specific treatment, which can shorten the duration of attacks, prevent complications, reduce the cost of treatment, and reduce the mortality rate.
Keywords: Porphobilinogen, Porphyria, Acute Intermittent, Female, Humans, Posterior Leukoencephalopathy Syndrome, Thailand, Hydroxymethylbilane Synthase, Phenotype
Background
Porphyria is a group of diseases that are related to a deficiency of enzymes in the heme synthesis pathway, which leads to accumulation of porphyrin precursors. The varied clinical presentations depend on each step of the enzyme defect. There are 2 main phenotypes: cutaneous and acute (hepatic) forms [1,2]. The true prevalence of porphyria is still unknown; however, it estimated to affect around 1 in 5500 to 5800 people in the United States and Europe [2].
Acute intermittent porphyria (AIP) is a common form of acute porphyria and is caused by a deficiency of the porphobilinogen deaminase enzyme that converts porphobilinogen to hydroxymethylbilane, resulting in excess porphobilinogen and aminolevulinic acid [3]. The classic symptoms include severe unexplained abdominal pain accompanied by nausea, vomiting, or constipation; neurological attacks, such as epilepsy, loss of sensation, and weakness; psychiatric symptoms, such as agitation, depression, insomnia, psychosis, delusions, and hallucinations; autonomic disturbances, such as hypertension and tachycardia; and abnormal sodium homeostasis [4]. Nevertheless, these symptoms are nonspecific and can be mimicked by other diseases. An acute attack is usually triggered by stress, sleep deprivation, fasting, infections, some medications, and menstruation [4].
AIP is passed by autosomal dominant inheritance and classified as a rare genetic disease, with a prevalence of 1 in 185 000 symptomatic individuals with AIP in Europe [4,5]. It can be challenging to establish a diagnosis of AIP due to the nonspecific clinical criteria and rarity of the disease.
Hydroxymethylbilane synthase (
Here, we present a case of AIP in a female adolescent patient who presented with classic features of an acute attack in AIP. Fortunately, she finally received a definitive diagnosis by confirming a pathogenic variant in
Our study was approved by the Human Research Ethics Committee of the Faculty of Medicine, Thammasat University No. 1 (MTU-EC-SA-1-236/62). Written informed consent was obtained from each participant.
Case Report
A 14-year-old Thai girl of non-consanguineous healthy parents was previously in good health, except for a history of febrile seizure at the age of 2 years. She was born uneventfully at term, with a birth weight of 3100 g. She had normal growth and development. There was no family history of any medical illness.
Ten days prior to admission at our hospital, she developed intermittent left lower quadrant abdominal pain with nausea and vomiting 4 to 6 times a day. The vomitus was non-bilious content. Seven days prior, she was brought to a local hospital and received omeprazole, metoclopramide, and antacid suspension treatment for gastritis and dyspepsia. However, she did not respond to treatment, and her symptoms worsened: she still had intractable intermittent abdominal pain, nausea, and vomiting 5 to 6 times a day. Therefore, 3 days prior to her referral, she was admitted to a general hospital. Her vomitus was only saliva because she was unable to eat or drink. Gastritis with moderate dehydration was the diagnosis. The treatment included 5% dextrose normal saline infusion, ondansetron, hyoscine butylbromide, and metoclopramide. Two days prior to referral, she developed loose stools twice a day; therefore, ceftriaxone was given as empirical treatment for enteritis. Then, 1 day prior to being referred, she had 2 episodes of brief generalized tonic seizure, with around 1 min before regaining consciousness. She was noticed to have high blood pressure (130–150/100 mmHg) for the past 3 days while admitted. Therefore, she was referred to our university hospital for suspected hypertensive encephalopathy, accompanied by severe intermittent abdominal pain and vomiting. She denied any fever, skin rash, or functional change. She lost 5 kg during these 10 days.
From the physical examination at our hospital, her vital signs revealed high blood pressure of 157/113 mmHg, pulse rate of 132 beats/min, temperature of 37.6°C, and respiratory rate of 22 breaths/min. Her body weight was 40 kg, and her height was 153 cm. She had good consciousness. Only mild tenderness at the suprapubic and left lower quadrant was noticed. The face, heart, lungs, skin, and neurological examination were all normal.
The initial differential diagnosis included acute poststreptococcal glomerulonephritis, systemic lupus nephritis, thyrotoxicosis, and pheochromocytoma. Therefore, initial laboratory investigations were sent; the results are shown in Table 1. Unexpectedly, the abnormalities were only mild hyponatremia, hypophosphatemia, and hypomagnesemia, which were caused by renal loss. The brain magnetic resonance imaging of T2 with fluid-attenuated inversion recovery revealed evidence of posterior reversible encephalopathy syndrome (PRES; Figure 1). Abdominal magnetic resonance imaging revealed 2 lobulated arterial hyper-enhancing masses with radiate fibrous septate arising from the enhancing central scar at hepatic segment 4b and 7, which were compatible with focal nodular hyperplasia. Liver function tests and the alpha-fetoprotein level were within the reference range.
She received the treatment for hypertensive encephalopathy by nicardipine intravenous (i.v.) drip for 1 day, and was then switched to oral propranolol and amlodipine. Ceftriaxone was continually given for empirical treatment of enteritis, and esomeprazole was given for dyspepsia. Her blood pressure decreased to the upper normal range for her age (120–130/80–90 mmHg) on the second day after admission. However, her abdominal pain persisted and constipation was noticed since the last loose stool was 4 days ago; therefore, polyethylene glycol was given.
Five days after admission, her blood pressure was in the normal range (110–120/70–80 mmHg), her abdominal pain was intermittent, she had no fever, and the bacterial cultures for blood, urine, and stool were negative. She developed confusion with proximal muscle weakness. Her blood chemistry showed severe hyponatremia, with a sodium level of 114 mmol/L, accompanied by euvolumia and high serum osmolarity (275 mmol/kg), compared with urine osmolarity (254 mmol/kg). Therefore, syndrome of inappropriate antidiuretic hormone (SIADH) was confirmed in this case of abdominal pain and PRES, which led to suspected AIP at day 8 after admission. The urine color change after exposure to sunlight is shown in Figure 2. The biochemical tests revealed high urine porphobilinogen, urine aminolevulinic acid, and serum aminolevulinic acid dehydrogenase (Table 2). The treatment of 150 g of glucose infusion per day with 10% dextrose i.v., and dextrin 40 g per day added in drinking water was given on day 8 after admission, with the caution of volume restriction for SIADH. High Carbohydrate loading, at least 300 g per day, has been the guideline standard treatment of AIP, with the aim of inhibiting the heme synthesis pathway [4,8]. Apart from carbohydrate loading, other supportive treatments for abdominal pain, nausea, and vomiting were administered by opioid analgesic, ondansetron, and diazepam.
After treatment for AIP was given, her clinical signs improved (blood pressure 106–123/64–86 mmHg and heart rate 80–100 beats/min). There was no more abdominal pain, but the weakness remained, and she developed depressive mood on day 12. At that time, her serum sodium level was 134 mmol/L (normal) and other blood chemistry results were in the reference range. She was then discharged on day 14. Her muscle weakness and depressive mood lasted for another 3 weeks, before returning to normal at 8 weeks after the onset of the acute attack. Her trigger may have been the fasting 12 h per day that she had done 1 week prior to the onset of her symptoms.
A molecular genetic test was sent to establish the definitive diagnosis. The patient’s deoxyribonucleic acid (DNA) was sent to a commercial laboratory for a gene panel of acute hepatic porphyria (
Sanger sequencing was performed to confirm the pathologic variant in our patient and all her available relatives, including her parents, sister, and paternal grandparents, by using genomic DNA extracted from their blood (Purgene® DNA extraction kit). A specific primer was designed: forward 5’-cagccagctagagagggaaa-3’, and reverse 5’-ggggatgactgtaaggcaga-3’. The reference sequence NM_000190.3 of
She had another 1 to 2 acute attacks per year; however, prompt and appropriate treatment was given; therefore, the later episodes lasted for only a few weeks. However, her renal function declined until the stage of chronic kidney disease; her blood urea nitrogen and creatinine levels were 24 mg/dL and 1.9 mg/dL, respectively.
Discussion
This case report of our patient is the first report of confirmed molecular diagnosis of AIP in Thailand. She presented with classic and complete features of AIP, starting with abdominal pain and vomiting, then developed hypertensive encephalopathy, followed by SIADH, psychiatric issues, and muscle weakness. If the prevalence of symptomatic AIP is around 1 in 200 000 individuals in the population [6], there are estimated to be more than 350 patients in Thailand. However, based on our personal communication with all Thai geneticists and researching all online publications, in which only biochemical tests were used for diagnosis, we determined that AIP has been diagnosed in fewer than 5 patients [9–11]. It remains difficult to infer whether the prevalence of AIP in Thai people is lower than that in Europeans or if the diagnosis of AIP has not been made.
Because AIP has multiple manifestations as neurovisceral symptoms, nearly all patients with acute attacks usually present with serious abdominal pain. Several cases were diagnosed with an acute surgical condition; however, there were negative surgical findings in these operations [10,12,13]. Other diagnoses that can mimic AIP include systemic lupus erythematosus or autoimmune disease, as in our patient, Guillain-Barré syndrome [14], inflammatory bowel disease [15], neurodegenerative diseases, and psychosis [16].
Hypertension and tachycardia are also classic symptoms of an autonomic disturbance, which can be distinguished from Cushing effect as a sign of increased intracranial hypertension. PRES has been reported in fewer than 40 AIP patients with acute attacks. More than 90% were in female patients and were associated with seizure, as in our patient. PRES in AIP might not be caused by hypertensive encephalopathy but can be from an accumulation of ALA, a neurotoxic substance [17–19].
Liver manifestations of AIP include elevation of serum aminotransferase, particularly in patients who have recurrent attacks [20]. Additionally, the risk of hepatocellular carcinoma is more common in symptomatic patients over the age of 60 years. However, the liver mass in our patient was compatible with focal nodular hyperplasia, which was an incidental finding. The treatment would be conservative, with clinical follow-up, in asymptomatic patients.
Currently, there have been more than 528 pathogenic/likely pathogenic variants reported in
A series of genotype p.Arg173Trp demonstrated more penetrance intra-family (up to 38%), more recurrent attacks with longer duration, more impaired renal function, and higher mortality than other genotypes [7,26]. The clinical manifestations of Arg173Trp variant in attacks are compared in Table 3. One patient with this genotype had frequent recurrent attacks until requiring liver transplantation for her treatment [27].
For a disease with such a low penetrance, although there is an inherited pathogenic variant in
The hemin infusion is a specific treatment for AIP, with efficacy in more than 85% of cases [28], resulting in faster resolution, lower incidence of complications, and shorter hospitalization, especially when given in the early stage of a moderate to severe acute attack [4,29]. The mechanism is suppressed alpha-aminoleuvunic acid synthase, whereby the level of aminoleuvunic acid and porphobilinogen are decreased. However, hemin is not available in several countries, including Thailand. Although, it can be ordered from abroad, the cost and duration of shipping are barriers. Hemin is also beneficial for prophylaxis episodes in frequent recurrent attacks, classified as 4 or more attacks that required hospitalization in 1 year [4]. The efficacy in recurrent attacks was found to be 68% [28]; however, thrombotic complications and iron overload needed to be monitored.
The prognosis of acute attacks is good, with most patients with AIP fully recovering; however, 5% to 20% of patients might have morbidity and mortality, especially in undiagnosed, delayed, or inappropriate treatment [29]. The long-term sequel-ae of AIP can be hepatocellular carcinoma, hypertension, and chronic kidney disease. Tubulointerstitial lesions are the main mechanism of renal disease, and rising creatinine levels are observed after an AIP attack; therefore, the recommendations are monitoring renal function, avoiding recurrent attacks, and nephrotoxic medications [29]. Hepatocellular carcinoma was observed with a prevalence of 21% to 27% in symptomatic and latent AIP [29,30]. Therefore, the recommendation should be screening by hepatic imaging (ultrasound) at 6-month intervals for early detection of liver cancer, especially in patients over 50 years old [31].
We would like to raise the awareness of considering AIP in the differential diagnosis in patients with multiorgan symptoms. The delay in diagnosis can cause over investigating and non-specific treatment, which may not be effective and can lead to lethal complications.
Conclusions
Because AIP is considered a rare disease due to its low incidence and varying symptoms and manifestations that make diagnosis challenging, this case report is aimed to improve the awareness of medical practitioners. Earlier diagnosis of AIP leads to prompt and specific treatment that can shorten the duration of attacks, prevent complications, lower the cost of treatment, and reduce the mortality rate.
Figures
Reference:
1.. Desnick RJ, Balwani M, The porphyria: Harrison’s principle of internal medicine, 2018; 2984-97, New York, McGraw-Hill Education
2.. Ramanujam VMS, Anderson KE, Porphyria diagnostics – part 1: A brief overview of the porphyrias: Curr Protoc Hum Genet, 2015; 86; 20.1-26 17
3.. Whatley SD, Badminton MN, Acute intermittent porphyria: GeneReviews® [Internet] Sep 27, 2005; 1993-2022, Seattle (WA), University of Washington Seattle [Updated 2019 Dec 5].
4.. Stein PE, Badminton MN, Rees DC, Update review of the acute porphyrias: Br J Haematol, 2017; 176; 527-38
5.. Elder G, Harper P, Badminton M, The incidence of inherited porphyrias in Europe: J Inherit Metab Dis, 2013; 36; 849-57
6.. Chen B, Solis-Villa C, Hakenberg J, Acute intermittent porphyria: Predicted pathogenicity of HMBS variants indicates extremely low penetrance of the autosomal dominant disease: Hum Mutat, 2016; 37; 1215-22
7.. Mvuz Fraunberg, Pischik E, Udd L, Kauppinen R, Clinical and biochemical characteristics and genotype-phenotype correlation in 143 Finnish and Russian patients with acute intermittent porphyria: Medicine (Baltimore), 2005; 84; 35-47
8.. Bustad HJ, Kallio JP, Vorland M, Acute intermittent porphyria: An overview of therapy developments and future perspectives focusing on stabilisation of HMBS and proteostasis regulators: Int J Mol Sci, 2021; 22; 675
9.. : Porphyria laboratory diagnostics in Thailand [Internet], 2019 [cited 2022 May 8]. Available from: https://royalsociety.go.th/porphyria-laboratory-diagnostics-in-thailand/
10.. Siriwong S, Acute porphyria attack after anesthesia: A case report: MJSBH, 2010; 26; 499-505
11.. Wiwanitkit V, Ehrlich reagent and diagnosis of acute porphyria: A note from Thailand: HAEMA, 2004; 7; 463-65
12.. Indika NLR, Kesavan T, Dilanthi HW, Many pitfalls in diagnosis of acute intermittent porphyria: A case report: BMC Res Notes, 2018; 11; 552
13.. Gouya L, Ventura P, Balwani M, EXPLORE: A prospective, multinational, natural history study of patients with acute hepatic porphyria with recurrent attacks: Hepatology, 2020; 71; 1546-58
14.. Mutluay B, Köksal A, Çelık RGG, Bülbül HH, A Case of acute intermittent porphyria mimicking Guillain-Barré syndrome: Noro Psikiyatr Ars, 2019; 56; 311-12
15.. Sieg I, Beckh K, Kersten U, Doss MO, Manifestation of acute intermittent porphyria in patients with chronic inflammatory bowel disease: Z Gastroenterol, 1991; 29; 602-5
16.. Crimlisk HL, The little imitator – porphyria: A neuropsychiatric disorder: J Neurol Neurosurg Psychiatry, 1997; 62; 319-28
17.. Zheng X, Liu X, Wang Y, Acute intermittent porphyria presenting with seizures and posterior reversible encephalopathy syndrome: Two case reports and a literature review: Medicine (Baltimore), 2018; 97; e11665
18.. Jaramillo-Calle DA, Solano JM, Rabinstein AA, Bonkovsky HL, Porphyria-induced posterior reversible encephalopathy syndrome and central nervous system dysfunction: Mol Genet Metab, 2019; 128; 242-53
19.. Swart G, Lim SS, Jude M, Acute intermittent porphyria presenting with posterior reversible encephalopathy syndrome (PRES) and abdominal pain: Pract Neurol, 2020; 20; 486-88
20.. Stein JA, Tschudy DP, Acute intermittent porphyria. A clinical and biochemical study of 46 patients: Medicine (Baltimore), 1970; 49; 1-16
21.. Gonzalez-Mosquera LF, Sonthalia S, Acute intermittent porphyria. [Updated 2022 May 8]: StatPearls [Internet] Jan, 2022, Treasure Island (FL), StatPearls Publishing [cited 2002 May 10]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK547665/
22.. Nunnemann S, Uibel C, Budig P, Mäurer M, Acute intermittent porphyria (AIP) in a patient with celiac disease: Neurol Res Pract, 2020; 2; 2
23.. Greene-Davis ST, Neumann PE, Mann OE, Detection of a R173W mutation in the porphobilinogen deaminase gene in the Nova Scotian “foreign Protestant” population with acute intermittent porphyria: A founder effect: Clin Biochem, 1997; 30; 607-12
24.. Tomie Y, Horie Y, Tajima F, Mutation in the exon 10 (R173W) of the hydroxymethylbilane synthase gene in two unrelated Japanese families with acute intermittent porphyria: Res Commun Mol Pathol Pharmacol, 1998; 99; 5-15
25.. Chen MC, Chang CJ, Lu YH, R173W mutation of hydroxymethylbilane synthetase is associated with acute intermittent porphyria complicated with rhabdomyolysis: The first report: J Clin Gastroenterol, 2015; 49; 256-57
26.. Andersson C, Floderus Y, Wikberg A, Lithner F, The W198X and R173W mutations in the porphobilinogen deaminase gene in acute intermittent porphyria have higher clinical penetrance than R167W. A population-based study: Scand J Clin Lab Invest, 2000; 60; 643-48
27.. Yasuda M, Erwin AL, Liu LU, Liver transplantation for acute intermittent porphyria: Biochemical and pathologic studies of the explanted liver: Mol Med, 2015; 21; 487-95
28.. Anderson KE, Collins S, Open-label study of hemin for acute porphyria: Clinical practice implications: Am J Med, 2006; 119; 801.e19-24
29.. Pischik E, Kauppinen R, An update of clinical management of acute intermittent porphyria: Appl Clin Genet, 2015; 8; 201-14
30.. Neeleman RA, Wagenmakers MAEM, Koole-Lesuis RH, Medical and financial burden of acute intermittent porphyria: J Inherit Metab Dis, 2018; 41; 809-17
31.. Balwani M, Wang B, Anderson KE, Acute hepatic porphyrias: Recommendations for evaluation and long-term management: Hepatology, 2017; 66; 1314-22
Figures
Tables
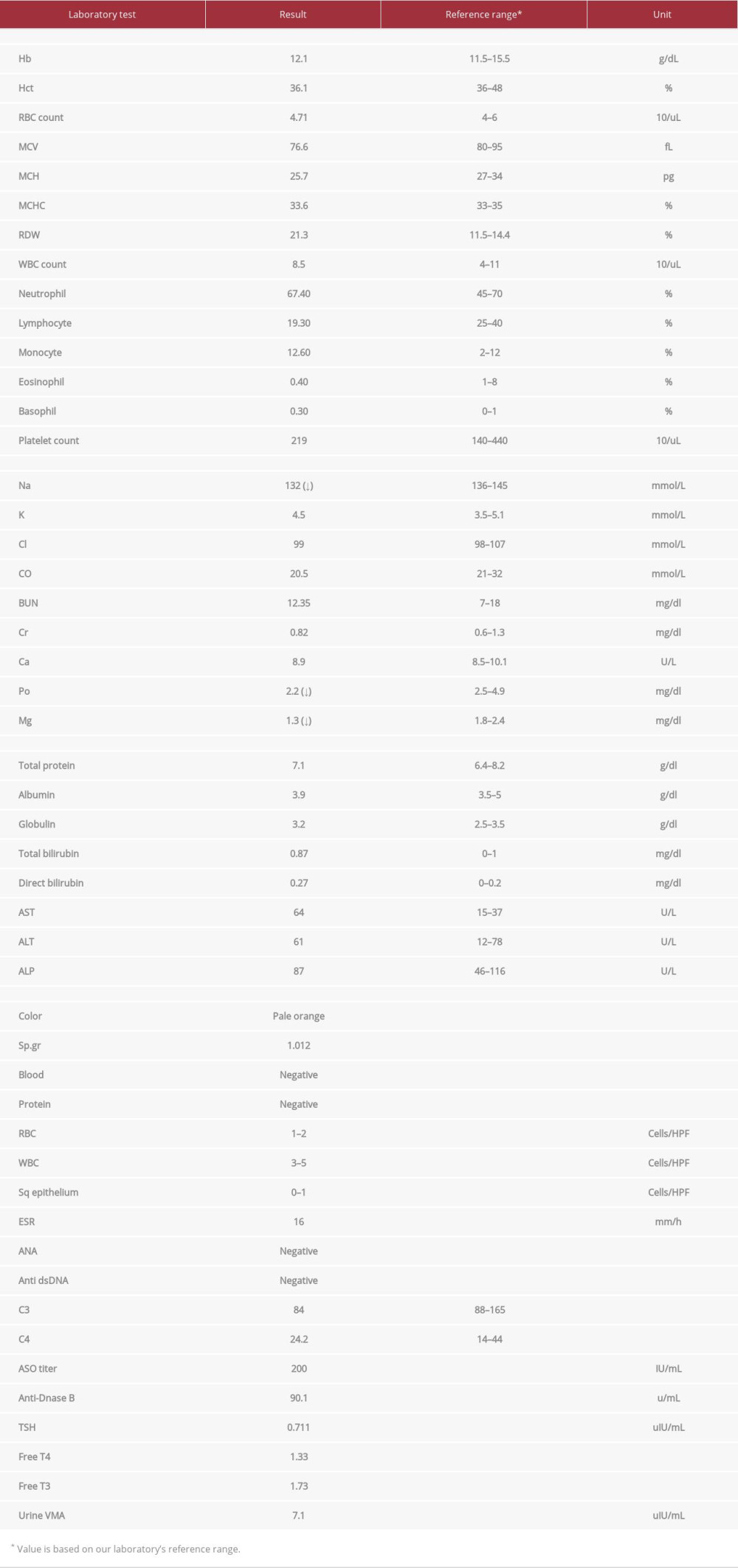 Table 1.. Initial laboratory results.
Table 1.. Initial laboratory results.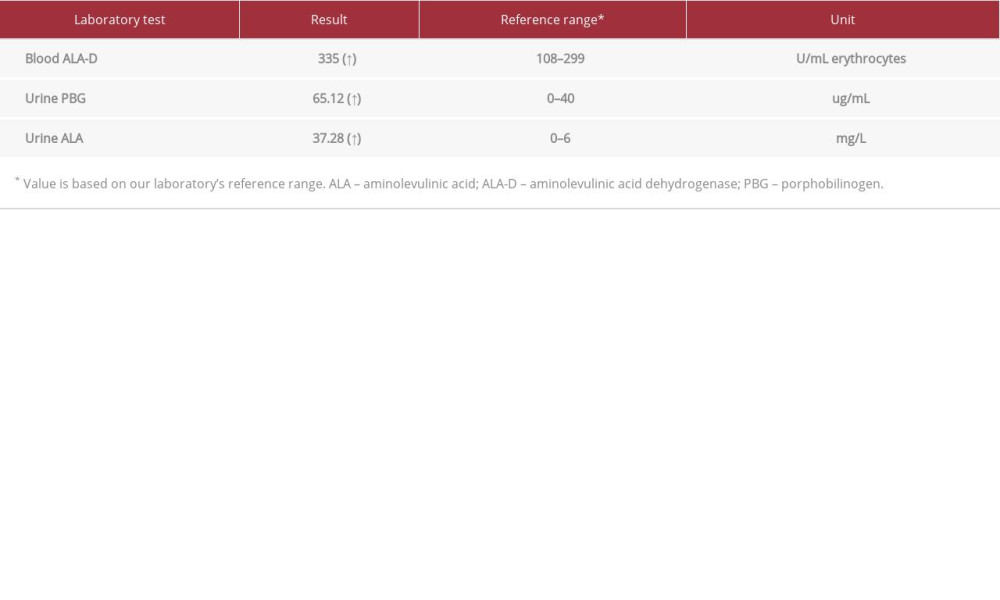 Table 2.. Specific blood results.
Table 2.. Specific blood results.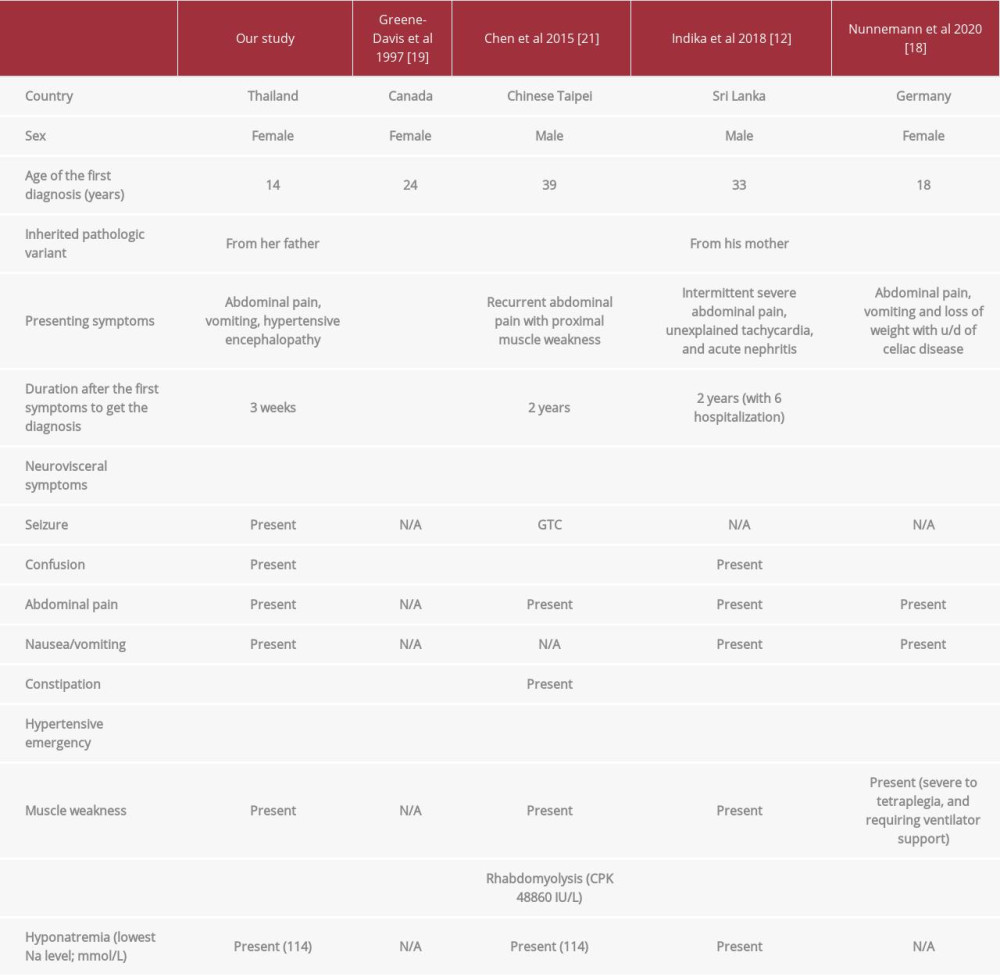 Table 3.. Comparing clinical manifestation of Arg173Trp variants in an acute attack.Table 1.. Initial laboratory results.Table 2.. Specific blood results.Table 3.. Comparing clinical manifestation of Arg173Trp variants in an acute attack.
Table 3.. Comparing clinical manifestation of Arg173Trp variants in an acute attack.Table 1.. Initial laboratory results.Table 2.. Specific blood results.Table 3.. Comparing clinical manifestation of Arg173Trp variants in an acute attack. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133