11 February 2023: Articles 
Mayer-Rokitansky-Küster-Hauser Syndrome with Situs Inversus Totalis: A Rare Case Report
Challenging differential diagnosis, Rare disease, Congenital defects / diseases, Rare coexistence of disease or pathology
Hari Soekersi1ACDEF, Riza Putri Aulia Hernowo1ACDEF*DOI: 10.12659/AJCR.939011
Am J Case Rep 2023; 24:e939011






Abstract
BACKGROUND: Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome is a complex disorder of the female reproductive system that results in an absent uterus and vagina. MRKH syndrome can be an isolated anomaly (typical) or accompanied by other organ anomalies (atypical). Due to the similarity of symptoms with other congenital gynecological diseases, imaging modalities remain the most important tools in establishing the diagnosis by visualizing internal genital and detecting possible organ malformations.
CASE REPORT: We present a very rare case of a female with primary amenorrhea. Pelvic magnetic resonance imaging (MRI) showed the absence of a uterus and vagina with possible Mullerian remnants, as well as an incidental finding of a right ectopic kidney. Abdominal ultrasonography and chest X-ray showed that the patient also had situs inversus totalis.
CONCLUSIONS: MRKH syndrome may be associated with situs inversus totalis due to possible early embryologic malformations causing both conditions; however, the exact mechanism is still unknown. This report should serve as a more recent attempt to question whether situs inversus totalis is related to MRKH and to emphasize the importance of imaging modalities, especially MRI, in establishing the diagnosis of MRKH syndrome and the associated malformations.
Keywords: Magnetic Resonance Imaging, Mullerian aplasia, Situs Inversus, Female, Humans, 46, XX Disorders of Sex Development, Uterus, Vagina, Dextrocardia
Background
Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome is characterized by congenital underdevelopment or absence of the uterus and upper two-thirds of the vagina in 46XX karyotypic females [1–5]. This malformation complex is a rare form of a Mullerian anomaly, with an incidence of 1 in 4000 to 5000 female live births [2,3,6]. Despite the rarity, it is a common cause of Mullerian anomalies and the second most frequent cause of primary amenorrhea [2]. In spite of malformation of the Mullerian ducts, the ovaries are often normal. The functioning ovaries result in the normal development of external genitalia and secondary sex characteristics, which makes the diagnosis quite difficult and often delayed until puberty, when primary amenorrhea occurs [3–6].
Situs inversus is a rare anomaly characterized by the mirror-image transposition of intraabdominal organs, relative to situs solitus [7]. Situs inversus is commonly accompanied by dextrocardia, or situs inversus totalis, when the location of the heart and its viscera is also mirrored [7]. Situs inversus develops due to an underlying genetic defect occurring in the early embryological development and thus can be associated with multiple congenital anomalies [8]. Early and thorough recognition of situs inversus totalis is important in establishing a diagnosis and treatment procedure, especially in emergency cases [7]. In this report, we present a case of MRKH syndrome with situs inversus totalis, as well as renal and skeletal anomalies, and the modalities used to confirm the diagnosis. To the best of our knowledge, this is a rare form of MRKH syndrome and only 3 cases have been reported in the literature.
Case Report
A 30-year-old woman was referred for ultrasound examination for primary amenorrhea. The patient never had any radiologic examinations before owing to a low economic status. The patient had begun pubertal development at 12 years of age and thelarche at 11 years. The patient did not report having cyclic pelvic pain. There was no family history of a similar problem. The patient’s mother did not use any medications that are known to be teratogenic, and there was no known radiation exposure during pregnancy. Hormonal analysis was within the normal range, and the karyotype was 46XX.
At physical examination, the patient was of average height and weight and had a normal arm span. The secondary sexual characteristics (breast, axillary, and pubic hair) were well developed (Tanner stage 5), with grossly normal external female genitalia. A rectal examination yielded no abnormality. The patient never had sexual intercourse before, and thus vaginal examination was not performed.
During transabdominal ultrasonography, an axial image showed the absence of normal uterine structure and the sagittal image showed no vagina between the urethra and anterior rectal wall (Figure 1). However, there was a small echoic structure at the posterior border of the bladder, which required further assessment. Abdominal ultrasonography images suggested that the patient had situs inversus (Figure 2). A chest X-ray showed that the heart was facing right and there was mild thoracal scoliosis (Figure 3).
A pelvic MRI was then performed, with contrast inserted via a urinary catheter to the bladder. The coronal T2 image identified both normal right and left ovaries, with volumes of 3.02 and 7.56 mL, respectively. Coronal and axial T2 images confirmed the absence of a uterus, but it showed 2 rudimentary horns with no endometrial cavity in the lateral pelvic wall, caudal to the ovaries, connected with a fibrous band (Figure 4A, 4B). The sagittal T2 image showed a triangular midline soft tissue structure and confirmed the complete vaginal agenesis (Figure 4C). An abdominal computed tomography scan and MRI were not performed; however, it could be seen from the coronal fat-suppressed T2 image that there was an ectopic right kidney in the pelvis (Figure 4D). From the karyotype test and imaging findings, MRKH was diagnosed, and the patient was given counseling regarding her condition.
Discussion
MRKH syndrome is mainly characterized by (1) malformation of the uterus and upper two-thirds of the vagina, (2) normal external genitalia, (3) functional ovaries, and (4) female karyo-type 46XX [1,9]. The exact cause of MRKH is still unknown, but it is believed to be polygenic and multifactorial [3]. In MRKH, there is a partial or complete failure of the Mullerian duct (primordia for the female internal reproductive systems) development prior to the sixth gestational week, which is believed to be related to the activated mutation of genes responsible for the anti-mullerian hormone or its receptor [6].
MRKH has 2 subtypes: type I (typical), characterized by isolated symmetrical uterovaginal aplasia or hypoplasia, and type II (atypical), characterized by Mullerian anomalies associated with other organ malformations [3,4,10]. The associated malformations in type II include urologic abnormalities (renal agenesis or hypoplasia, renal ectopic, hydronephrosis, or horseshoe kidney), skeletal abnormalities (ribs, extremities, and especially spine, such as Klippel-Feil anomaly, fused vertebrae, and scoliosis), auditory problems (middle ear malformations, sensorineural defects, and auditory meatus adysplasia), and, rarely, cardiac abnormalities (aorto-pulmonary window, atrial septal defects, pulmonary valvular stenosis, and Tetralogy of Fallot) [4,10].
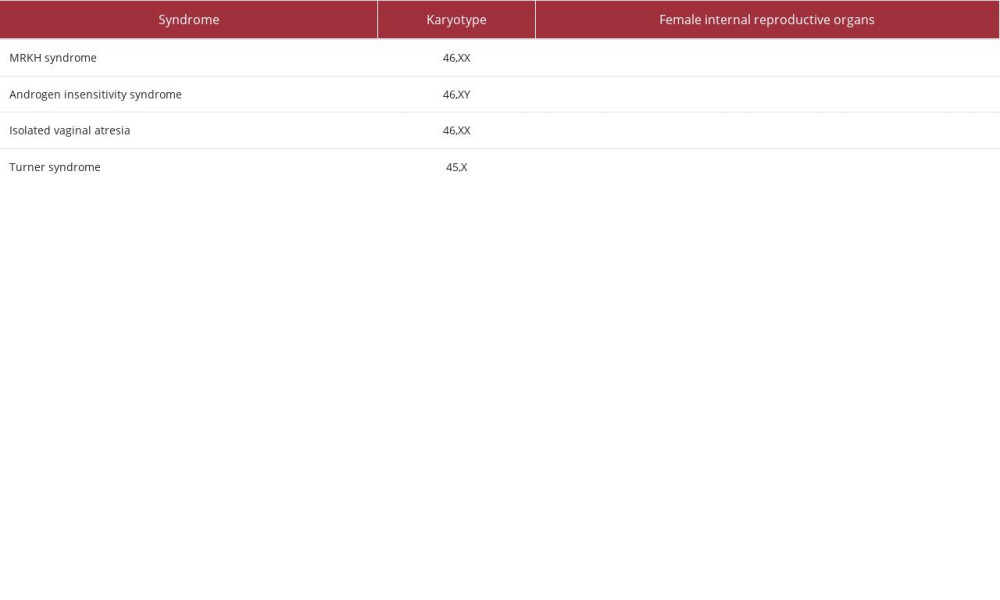
Patients with MRKH have a 46XX karyotype; thus, the levels of follicle stimulating hormone and luteinizing hormone are normal due to normally functioning ovaries [2–4,11]. These normal hormones can cause similar symptoms as other gynecological diseases, such as cyclic abdominal pain due to developed endometrial tissue [1], as well as cyclical breast and mood alterations, consistent with ovulations [6]. Differential diagnosis of MRKH includes diseases with symptoms of primary amenorrhea but normal secondary sexual characteristics (Table 1) [1,3,5,12,13].
Diagnosis of MRKH syndrome is established after no visualization of a normal uterus and of the upper vagina, even though the ovaries are normal [5,14]. Of all available imaging modalities, MRI is the criterion standard for providing detailed morphology of the internal genital organs and accurately determining the uterine aplasia, as well as for depicting any coexisting anatomy and providing a road map for surgical planning [5,11,12,15]. MRI is superior to other modalities due to its safety, lack of ionizing radiation, and higher soft-tissue contrast resolution in superficial and deep planes [3,4,6,14,15]. In study by Fedele et al, MRI was found to give more detailed information than ultrasonography and laparoscopy, which was presumed to be from the lack of ability of laparoscopy to visualize sub-peritoneal organs and the superiority of MRI to evaluate the endometrial stripe [16]. While laparoscopy is required in some cases, MRI is still useful in postponing the exploration by providing initial assessment of the pelvis [14].
Even though uterine absence is a rule in MRKH syndrome, there can be remnants of the Mullerian duct, which mainly present as rudimentary uterine buds, along with fibrous band-like structures connecting the buds and midline triangular soft tissue lying over the bladder dome [3,9,14]. According to Pompili, more than 50% of patients present with bilateral buds while the rest have a unilateral bud or no bud at all [14]. Uterine buds are usually located antero-inferior to their respective ipsilateral ovaries [17] and they appear as a dark solid ovoid structure on T1-weighted imaging and can appear cavitated with a target-sign appearance on T2-weighted imaging [14,18]. Identifying these remnants is important owing to the higher chance of creating a functioning uterus [14,17] or for the possibility of further issues arising from the remaining functional endome-trial tissues, such as endometriosis or hematometra [3,6,14].
In addition to assessing the uterus and Mullerian remnants, MRI is also used to assess the vagina, ovaries, and other organ malformations that can occur in patients with MRKH syndrome. Even though the lower two-thirds of the vagina has different embryogenesis than the upper one affected by MRKH, some patients can have a completely absent vagina while others have a short vaginal pouch or indentation superior to the hymenal ring [19]. The presence of a remaining vagina should be carefully assessed for future treatment, especially its length from the urethral meatus to influence the option for vaginal canal formation [5,14]. Ovaries in MRKH syndromes are present; however, they can be located in extra-pelvic locations [5]. Reporting the location of the ovaries is necessary to prevent any complications, such as torsion, incarceration, or ischemia [20], as well as to guide clinicians in choosing an abdominal or transvaginal route in ovum retrieval during in vitro fertilization or surrogacy process [18].
In this report, MRI of our patient showed the absence of normal structure of the uterus and confirmed the presence of a thick fibrous band extending from the bilateral uterine buds and converging onto a midline triangular, which is presumed to represent the Mullerian duct remnants in MRKH containing utero-ovarian and round ligaments [17]. The sagittal image also showed complete vaginal agenesis, which is possible in MRKH if there is a complete arrestment of the paramesonephric ducts [11]. Pelvic MRI also showed the presence of an ectopic right kidney, which is in accordance with previous studies that showed renal abnormalities are common in atypical MRKH due to the fact that both renal and reproductive systems originate from the mesodermal mass in the same embryological period [4,10,21]. Even though no laparoscopy was performed in this patient because she had no indication for mass or remnant removal, MRI was considered enough to diagnose MRKH due to its ability to detect the genital organ malformations.
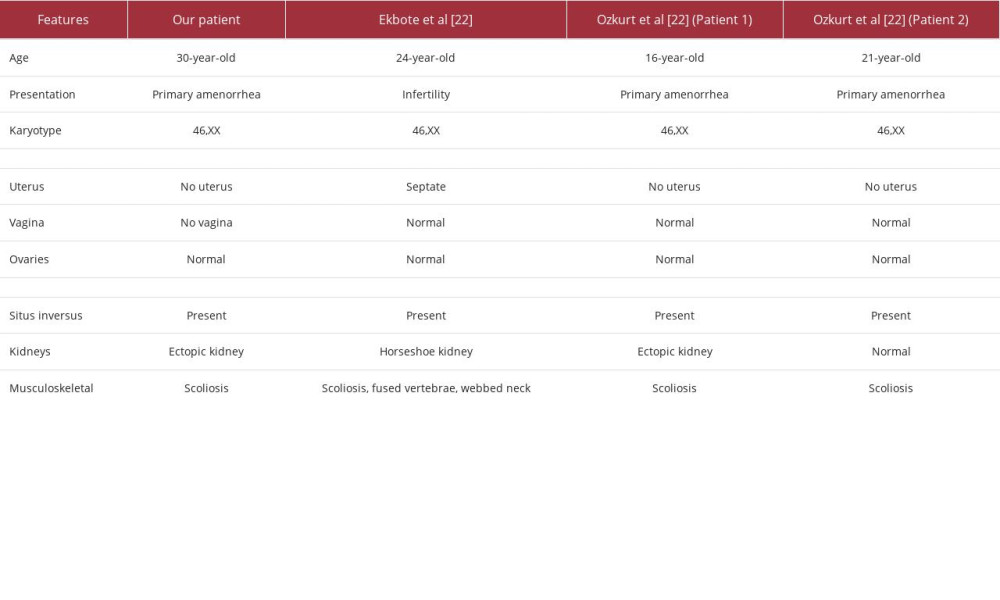
Other than MRKH syndrome, our patient’s radiologic examinations suggested that the patient had situs inversus totalis; this coexistence has been stated in only 3 patients [22,23]. The comparison of the patients is presented in Table 2. Scoliosis and renal anomalies are also found in other patients, which confirmed the involvement of skeletal and genitourinary malformations in the syndrome. The association of MRKH and skeletal and genitourinary malformations may be due to alterations happening in the end of the fourth gestational week, as there is a close spatial relationship of the blastemata of the lower cervical and upper thoracic somites, as well as the pronephrotic ducts [24]. The exact mechanism for situs inversus totalis and MRKH is still unknown; however, it is possible that early dysgenesis leading to situs inversus may also result in other organ malformations, including of the reproductive systems.
The complete absence of a uterus and vagina narrowed the treatment options of our patient to a surgical approach to create a neovagina or resect the Mullerian remnants if needed. The findings of inverted organs and ectopic kidney in our patient may assist clinicians in determining the surgical approach as some techniques may be considered safer than the others due to lower risk of ureteral injury [14,17].
Conclusions
In conclusion, we report an extremely rare case of MRKH syndrome with situs inversus totalis. This report should serve as a more recent attempt to question whether total situs inversus is related with MRKH and to emphasize the importance of MRI in diagnosing MRKH and the associated malformations.
Figures
References:
1.. Giusti S, Fruzzetti E, Perini D, Diagnosis of a variant of Mayer-Rokitansky-Kuster-Hauser syndrome: Useful MRI findings: Abdom Imaging, 2011; 36(6); 753-55
2.. Govindarajan M, Rajan RS, Kalyanpur A, Ravikumar , Magnetic resonance imaging diagnosis of Mayer-Rokitansky-Kuster-Hauser syndrome: J Hum Reprod Sci, 2008; 1(2); 83-85
3.. Kara T, Acu B, Beyhan M, Gökçe E, MRI in the diagnosis of Mayer-Rokitansky-Kuster-Hauser syndrome: Diagn Interv Radiol (Ankara, Turkey), 2013; 19(3); 227-32
4.. Morcel K, Camborieux L, Guerrier D, Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: Orphanet J Rare Dis, 2007; 2; 13
5.. Rousset P, Raudrant D, Peyron N, Ultrasonography and MRI features of the Mayer-Rokitansky-Küster-Hauser syndrome: Clin Radiol, 2013; 68(9); 945-52
6.. Folch M, Pigem I, Konje JC, Müllerian agenesis: Etiology, diagnosis, and management: Obstet Gynecol Surv, 2000; 55(10); 644-49
7.. Fulcher AS, Turner MA, Abdominal manifestations of situs anomalies in adults: Radiographics, 2002; 22(6); 1439-56
8.. Abut E, Arman A, Güveli H, Malposition of internal organs: A case of situs ambiguous anomaly in an adult: Turk J Gastroenterol, 2003; 14(2); 151-55
9.. Yoo RE, Cho JY, Kim SY, Kim SH, Magnetic resonance evaluation of Müllerian remnants in Mayer-Rokitansky-Küster-Hauser syndrome: Korean J Radiol, 2013; 14(2); 233-39
10.. Oppelt P, Renner SP, Kellermann A, Clinical aspects of Mayer-Rokitansky-Kuester-Hauser syndrome: Recommendations for clinical diagnosis and staging: Hum Reprod (Oxford, England), 2006; 21(3); 792-97
11.. Rogers BC, Merideth KL, Sonographic detection of Mayer-Rokitansky-Küster-Hauser syndrome: J Diagn Med Sonogr, 2015; 31(2); 103-8
12.. Fiaschetti V, Taglieri A, Gisone V, Mayer-Rokitansky-Kuster-Hauser syndrome diagnosed by magnetic resonance imaging. Role of imaging to identify and evaluate the uncommon variation in development of the female genital tract: J Radiol Case Rep, 2012; 6(4); 17-24
13.. Guerrier D, Mouchel T, Pasquier L, Pellerin I, The Mayer-Rokitansky-Küster-Hauser syndrome (congenital absence of uterus and vagina) – phenotypic manifestations and genetic approaches: J Negat Results Biomed, 2006; 5; 1
14.. Pompili G, Munari A, Franceschelli G, Magnetic resonance imaging in the preoperative assessment of Mayer-Rokitansky-Kuster-Hauser syndrome: Radiol Med, 2009; 114(5); 811-26
15.. Church DG, Vancil JM, Vasanawala SS, Magnetic resonance imaging for uterine and vaginal anomalies: Curr Opin Obstet Gynecol, 2009; 21(5); 379-89
16.. Fedele L, Dorta M, Brioschi D, Magnetic resonance imaging of unicornuate uterus: Acta Obstet Gynecol Scand, 1990; 69(6); 511-13
17.. Bhayana A, Ghasi RG, MRI evaluation of pelvis in Mayer-Rokitansky-Kuster-Hauser syndrome: Interobserver agreement for surgically relevant structures: British J Radiol, 2019; 92(1097); 20190045
18.. Hall-Craggs MA, Williams CE, Pattison SH, Mayer-Rokitansky-Kuster-Hauser syndrome: Diagnosis with MR imaging: Radiology, 2013; 269(3); 787-92
19.. Gould SW, Epelman M, Magnetic resonance imaging of developmental anomalies of the uterus and the vagina in pediatric patients: Semin Ultrasound CT MRI, 2015; 36(4); 332-47
20.. Boruah DK, Sanyal S, Gogoi BB, Spectrum of MRI appearance of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome in primary amenorrhea patients: J Clin Diagn Res, 2017; 11(7); TC30-35
21.. Willemsen WN, Renal- skeletal- ear- and facial-anomalies in combination with the Mayer--Rokitansky-Küster (MRK) syndrome: Eur J Obstet Gynecol Reprod Biol, 1982; 14(2); 121-30
22.. Ekbote AV, Kamath MS, Danda S, MURCS association with situs inversus totalis: Expanding the spectrum or a novel disorder: J Pediatr Genet, 2014; 3(3); 167-73
23.. Ozkurt H, Cenker MM, Keskiner F, Basak M, Two cases of Mayer-Rokitansky-Kuster-Hauser syndrome with situs inversus totalis: Coincidence or co-existence?: J Pediatr Adolesc Gynecol, 2009; 22(4); e57-60
24.. Braun-Quentin C, Billes C, Böwing B, Kotzot D, MURCS association: Case report and review: J Med Genet, 1996; 33(7); 618-20
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report  22,759,844
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133