30 August 2024: Articles 
Concurrent Gitelman Syndrome and Hyperthyroidism: Diagnostic Challenges in a 51-Year-Old Patient
Rare disease
Yan Zhang1AE, Hao Yu2BF, Jun Li1B, Ling ChengDOI: 10.12659/AJCR.944909
Am J Case Rep 2024; 25:e944909






Abstract
BACKGROUND: Gitelman syndrome (GS) is an uncommon autosomal recessive inherited disease caused by inactivating mutations in the SLC12A3 gene located on chromosome 16q13, resulting in distal tubular dysfunction. Most cases are detected during routine examinations in adulthood, due to hypokalemia and alkalosis. GS needs to be distinguished from diseases that cause hypokalemia, such as Classic Bartter syndrome and hyperthyroidism. In individual cases, GS and hyperthyroidism occur simultaneously, which is prone to misdiagnosis.
CASE REPORT: A 51-year-old woman with intermittent palpitations and lower limb fatigue for 4 years received a diagnosis of hypokalemia at a local hospital. Treatment with potassium supplementation did not improve the patient’s palpitations and fatigue. After coming to our hospital for examination, it was found that the patient had hyperthyroidism. After receiving treatment of hyperthyroidism remission and sufficient potassium replacement, the patient’s serum potassium level remained low. Meanwhile, the patient had hypomagnesemia and metabolic alkalosis. Subsequently, according to our suggestion, the patient continued to take oral supplements of potassium and magnesium, while also started on spironolactone. We convinced the patient to undergo genetic testing and discovered compound heterozygous mutations in the SLC12A3 gene, which presented a definitive diagnosis of GS. In the following 3 months, the patient’s serum potassium level was within the normal range, and the dose of methimazole was reduced.
CONCLUSIONS: As a rare disease, GS may have only mild or occasional manifestations, making it prone to misdiagnosis. GS remains therapeutically challenging, and future progress in treatment will depend on further research of the disease.
Keywords: Gitelman syndrome, hyperthyroidism, Hypokalemia, Humans, Middle Aged, Female, Diagnosis, Differential, Solute Carrier Family 12, Member 3
Introduction
Gitelman syndrome (GS) is a desalinated renal tubular disease characterized by hypokalemia and metabolic alkalosis [1,2]. The genetic mode of GS is autosomal recessive inheritance, caused by inactivation mutations in the gene SLC12A3 encoding the thiazide-sensitive sodium chloride cotransporter (NCCT) located in the distal tubules of the kidneys [1]. In 2019, GS was included in the first list of rare diseases in China. A common problem with rare diseases is the lack of high-quality evidence to guide treatment and predict prognosis, thereby requiring more relevant research and case reports.
In addition to GS, some endocrine disorders, including hyper-thyroidism, can cause hypokalemia. Hyperthyroidism is caused by the excessive synthesis and release of thyroid hormones by the thyroid gland, leading to metabolic hyperactivity and sympathetic nervous system excitation in the body. Although patients with GS rarely have hyperthyroidism, the presence of hyperthyroidism can easily lead to misdiagnosis and delay GS treatment.
We report a middle-aged Chinese woman who presented with hypokalemia and was found to have hyperthyroidism. After receiving remission of hyperthyroidism and adequate potassium replacement therapy, the patient’s serum potassium level remained below normal. Genetic testing confirmed the diagnosis of GS [1]. After treatment with potassium, magnesium supplementation, and oral spironolactone, the patient’s serum potassium level continued to be maintained within the normal range. We present this case in accordance with the CARE reporting checklist.
Case Report
In 2018, a 51-year-old Chinese woman received a diagnosis of hypokalemia at a local hospital, due to intermittent palpitations and fatigue. After receiving sufficient potassium supplementation treatment, the patient’s symptoms improved. In the previous 4 years, the patient had experienced intermittent palpitations and fatigue and had visited the local hospital multiple times, continuing potassium supplementation treatment. In January 2024, the patient sought medical attention at our hospital, due to frequent episodes of intermittent palpitations. The patient’s vital sign examination showed a heart rate of 73 beats per min and blood pressure of 113/72 mmHg. The patient had no history of diarrhea or vomiting and denied taking laxatives or diuretics. Upon review of the medical history, it was noted that the patient received local radiotherapy and immunotherapy for cervical cancer at a local hospital in 2022. The patient had 2 full-term pregnancies and delivered normally. Her parents and 2 sons had no history of hypokalemia or related symptoms. The patient’s family mentioned that the doctor at the local hospital had considered diagnosing Bartter syndrome during their previous visits. However, the patient ultimately refused to undergo genetic testing.
After admission, complete biochemical tests were conducted, and the results showed that the serum potassium level was 3.42 mmol/L, serum magnesium level was 0.44 mmol/L, and serum calcium level was 2.09 mmol/L (Table 1). To improve the patient’s symptoms, 3 g of potassium chloride sustained-release tablets and 2 tablets of potassium magnesium aspartate containing approximately 23.6 mg of magnesium were administered orally daily, and 10 mL of calcium gluconate was temporarily intravenously supplemented. Meanwhile, thyroid function examination revealed hyperthyroidism (thyroid stimulating hormone 0.06 μIU/mL, free triiodothyronine 8.16 pmol/L, free tetraiodothyronine 30 pmol/L). The thyroid antibodies, including anti-thyroid peroxidase antibody, antithyroglobulin antibody, and thyrotrophin receptor antibody, were all negative (Table 1). Cortisol and adrenocorticotropin were also in the normal range, at 8: 00 AM (Table 1). Although all thyroid antibodies were negative, low-dose anti-thyroid drug was considered, to improve the obvious symptom of palpitations. Therefore, the patient began taking 10 mg of methimazole orally daily. Subsequently, other test results showed that the patient’s blood gas analysis indicated metabolic alkalosis, and 24-h urine testing revealed that the levels of urinary calcium, urinary magnesium, and urinary phosphorus were all below the reference range, while the levels of renin at different body positions in the blood were elevated (Tables 1–3). After pausing potassium supplementation for 24 h, the urine potassium level was detected to be within the reference range (Table 3). The ultrasound examination of the thyroid and bilateral kidneys showed no significant abnormalities. Despite multiple communications with the patient and her family, the patient insisted on refusing to undergo genetic testing, and also refused to use spironolactone.
After taking anti-thyroid drug and supplementing with potassium and magnesium for 1 week, the patient underwent a follow-up examination. The serum potassium level was 2.66 mmol/L, serum magnesium level was 0.57 mmol/L, and calcium level was within the reference range (2.21 mmol/L). The patient’s symptom of intermittent palpitations had not completely improved. After repeated persuasion, the patient began taking spironolactone orally (10 mg twice daily). Monitoring of the patient’s blood pressure closely showed fluctuations from 95/68 mmHg to 115/72 mmHg. Considering the diuretic effect of spironolactone and the risk of causing hypotension, the dosage of spironolactone was not increased. After further discussion with the patient and her family, the patient ultimately agreed to undergo genetic testing. A compound heterozygous mutation of c.179C>T in exon1 and c.961C>T in exon7 of the SLC12A3 gene was found, confirming the diagnosis of GS (Figure 1). Therefore, the patient continued to receive potassium and magnesium supplements, oral spironolactone, and anti-thyroid treatment. The patient was followed up in the outpatient department, and the levels of potassium, magnesium, and calcium in blood were all controlled. At the same time, the patient reached a normal thyroid state, and the dosage of anti-thyroid drug was gradually decreased.
Discussion
GS, characterized by hypokalemia, metabolic alkalosis, hypomagnesemia, and hypocalciuria, is a rare hereditary disease [1].
The prevalence of GS ranges from 1 to 10 in 40 000, making it the most common hereditary renal tubular disease. The pathogenesis of GS is due to mutations in the SLC12A3 gene, which leads to the inactivation of NCCT encoded by it, causing impaired reabsorption of sodium and chlorine in the renal distal tubules, and subsequently leading to a series of clinical symptoms [1]. GS often occurs in adolescents or adults, and occasionally in children. GS has a hidden onset and diverse clinical manifestations, making it prone to misdiagnosis or missed diagnosis. Here, we report a 51-year-old woman who presented with hypokalemia and was found to have hyperthyroidism. After receiving remission of hyperthyroidism and adequate potassium replacement therapy, the patient’s serum potassium level remained below normal. Genetic testing revealed a compound heterozygous mutation in the SLC12A3 gene, which allowed for the diagnosis of GS. After 1 month of treatment with potassium, magnesium supplementation, and oral spironolactone, the patient’s serum potassium could be maintained within the reference range.
GS patients often seek medical attention due to symptoms such as fatigue and limb weakness caused by low potassium. The common clinical causes of hypokalemia include insufficient intake, increased excretion, and intracellular potassium transfer. Due to the absence of symptoms such as frequent vomiting, long-term diarrhea, and excessive sweating, hypokalemia caused by reduced intake and increased potassium excretion through the digestive tract and skin can be ruled out. In the present case, the patient detected hyperthyroidism and received treatment immediately. Excessive secretion of thyroid hormones can directly enhance the Na+- K+-ATPase activity of cells, leading to intracellular potassium ion transfer and subsequently causing hypokalemia. In reviewing previous literature, we found that GS can be associated with thyroid disease [3]. Almost all of these patients are from East Asian population, which cannot be excluded from the high incidence rate of autoimmune thyroid disease in this region. A study in Japan showed that approximately 4.3% of patients with GS have thyroid dysfunction [3]. Compared with other types of autoimmune thyroid disease, GS and Graves disease are more likely to coexist [4]. At present, there is no clear evidence to suggest that the SLC12A3 gene is associated with the pathogenesis of thyroid diseases [5]. At the same time, iodine metabolism is closely related to magnesium metabolism, and the low level of magnesium in the blood can lead to the recurrence of hyperthyroidism [6].
Unlike in other reported cases of GS combined with hyperthyroidism, our patient’s thyroid ultrasound showed no abnormalities and thyroid-related antibodies were negative. Meanwhile, an article analyzing thyroid function in 35 patients with GS suggests that thyroid dysfunction in patients with GS can be caused by factors other than autoimmune thyroid disease [7]. Among these GS patients, the serum magnesium levels of patients with hyperthyroidism were lower than those of patients with normal thyroid function. The use of methimazole leads to an increase in the level of serum magnesium. Nevertheless, no significant difference was found in the incidence of thyroid dysfunction between a hypomagnesemia group and a normal magnesemia group [7]. It is undeniable that the sample size of the present study is limited, and further confirmation is needed through studies with larger sample sizes. Looking back at the patient’s past history, more than a year ago, the patient received immune checkpoint inhibitor treatment for early cervical cancer. However, thyroid dysfunction caused by immune checkpoint inhibitors generally occurs within 1 to 2 months of starting treatment [8]. After the patient received small doses of anti-thyroid drug and supplementation with potassium and magnesium, her thyroid function quickly returned to the reference range. Therefore, we speculated that the patient’s hypomagnesemia may have been the cause of hyperthyroidism. Of course, more research is needed to elucidate the detailed mechanisms of thyroid dysfunction in patients with GS.
After the patient took the anti-thyroid drug and sufficient potassium supplementation for a week, the patient’s thyroid gland transitioned to a normal state, but the level of serum potassium remained below the reference range. Meanwhile, it was found that the patient had hypomagnesemia, low urinary calcium, and elevated renin levels. Therefore, GS or classic Bartter syndrome was highly suspected as a diagnosis. Bartter syndrome was first described by Dr. Frederick Barthes in 1962, and is characterized by hypokalemic alkalosis, elevated levels of renin and aldosterone but normal blood pressure, and proliferation and hypertrophy of the periglomerular organs [9]. Bartter syndrome is divided into 7 different types based on clinical manifestations. Among them, Bartter syndrome type III, also known as classic Bartter syndrome, affects about 20% to 30% of patients with Bartter syndrome and is a hereditary disease caused by mutations in the CLCNKB gene encoding the chloride ion channel CIC-Kb [9]. Unlike other types of Bartter syndrome, classic Bartter syndrome rarely presents with renal calcification and retains renal concentration ability. Although commonly considered a mild Bartter syndrome variant, patients with classic Bartter syndrome typically have the most severe electrolyte abnormalities. The clinical features of classic Bartter syndrome overlap most with GS, and they are both classified as diseases that cause dysfunction of the distal tubules of the kidney [1]. Compared with patients with GS, patients with classic Bartter syndrome have a relatively early onset (mostly before the age of 3 years), and are more prone to delayed growth and development, normal serum magnesium levels, and high urinary calcium levels. In some cases, patients can also experience occasional hypokalemia, hypomagnesemia, or hypocalciuria in later adulthood. With the increasing number of reported cases of GS and classic Bartter syndrome, we have found that GS and classic Bartter syndrome have many overlapping phenotypes, making diagnosis based solely on phenotype data challenging. The detection of SLC12A3 mutations with biallelic gene inactivation is crucial for the diagnosis of GS [1].
So far, more than 350 mutations distributed in SLC12A3 have been found in patients with GS [1]. The next-generation gene sequencing technology has become easier to obtain and more comprehensive, and is worth promoting in suspected patients with GS. However, the higher testing costs have led to some patients giving up on a clear diagnosis.
During the long-term management and treatment of GS, patients should be informed of the adverse effects of taking corresponding supplements. Common examples include abdominal pain and diarrhea caused by magnesium salts, as well as gastric irritation caused by potassium chloride [1]. Furthermore, potassium-sparing diuretics, renin-angiotensin system inhibitors, and nonsteroidal anti-inflammatory drugs have also been considered for use. Potassium-sparing diuretics are the most commonly used, with commonly used drugs including spironolactone and epinephrine. These drugs can increase serum potassium levels in patients who are resistant to supplements, as well as treat magnesium consumption that worsens due to elevated aldosterone levels [9]. Spironolactone is limited in use among adolescents and young people due to its anti-androgenic effects, such as male breast development, erectile dysfunction, and menstrual irregularities [10]. Eprolidone is a selective aldosterone antagonist that has no anti-androgenic adverse effects, compared with spironolactone [10]. During the use of these drugs, blood pressure needs to be monitored to avoid hypotension. There have been reports of using reninangiotensin system inhibitors in the treatment of GS. Due to its long-term cardiovascular effects, its widespread promotion is limited [1]. Considering the short-term and long-term gastrointestinal adverse effects and nephrotoxicity of nonsteroidal anti-inflammatory drugs, caution should be exercised when using them [11].
So far, there is no evidence to suggest that GS will affect life expectancy. As a rare disease, GS lacks large-scale clinical studies and requires long-term research to evaluate its natural history and potential complications in various systems.
Conclusions
GS is a rare autosomal disease, with atypical clinical manifestations in most cases. Many non-hereditary diseases can mimic GS expression. Therefore, GS is easily overlooked or misdiag-nosed, which further emphasizes the importance and necessity of molecular diagnosis for GS. The management of GS should be individualized, while monitoring the potential complications and evolution. Further research is needed on the relevant cases in order to better summarize the treatment experience.
References:
1.. Blanchard A, Bockenhauer D, Bolignano D, Gitelman syndrome: Consensus and guidance from a kidney disease: Improving Global Outcomes (KDIGO) Controversies Conference: Kidney Int, 2017; 91(1); 24-33
2.. Simon DB, Nelson-Williams C, Bia MJ, Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter: Nat Genet, 1996; 12(1); 24-30
3.. Fujimura J, Nozu K, Yamamura T, Clinical and genetic characteristics in patients with Gitelman syndrome: Kidney Int Rep, 2018; 4(1); 119-25
4.. Qin YZ, Liu YM, Wang Y, Novel compound heterozygous mutation of SLC12A3 in Gitelman syndrome co-existent with hyperthyroidism: A case report and literature review: World J Clin Cases, 2022; 10(21); 7483-94
5.. Koca O, Alay MT, Murt A, A novel homozygous SLC12A3 mutation causing Gitelman syndrome with co-existent autoimmune thyroiditis: A case report and review of the literature: CEN Case Rep, 2024 [Online ahead of print],
6.. Xu J, He J, Xu S, Gitelman syndrome with Graves’ disease leading to rhabdomyolysis: A case report and literature review: BMC Nephrol, 2023; 24(1); 123
7.. Zhou H, Ren Y, Lu C, Thyroid function in 35 patients with Gitelman syndrome: Biomed Res Int, 2020; 2020; 7963898
8.. Iwama S, Kobayashi T, Yasuda Y, Arima H, Immune checkpoint inhibitor-related thyroid dysfunction: Best Pract Res Clin Endocrinol Metab, 2022; 36(3); 101660
9.. Fulchiero R, Seo-Mayer P, Bartter syndrome and Gitelman syndrome: Pediatr Clin North Am, 2019; 66(1); 121-34
10.. Huang X, Wu M, Mou L, Gitelman syndrome combined with diabetes mellitus: A case report and literature review: Medicine (Baltimore), 2023; 102(50); e36663
11.. Peng X, Chen C, Tu J, Long-term indomethacin treatment in a Chinese child with Gitelman syndrome: Case report and literature review on its efficacy and tolerance: Am J Case Rep, 2023; 24; e941627
Tables
 Table 1.. Laboratory findings of the patient on admission.
Table 1.. Laboratory findings of the patient on admission.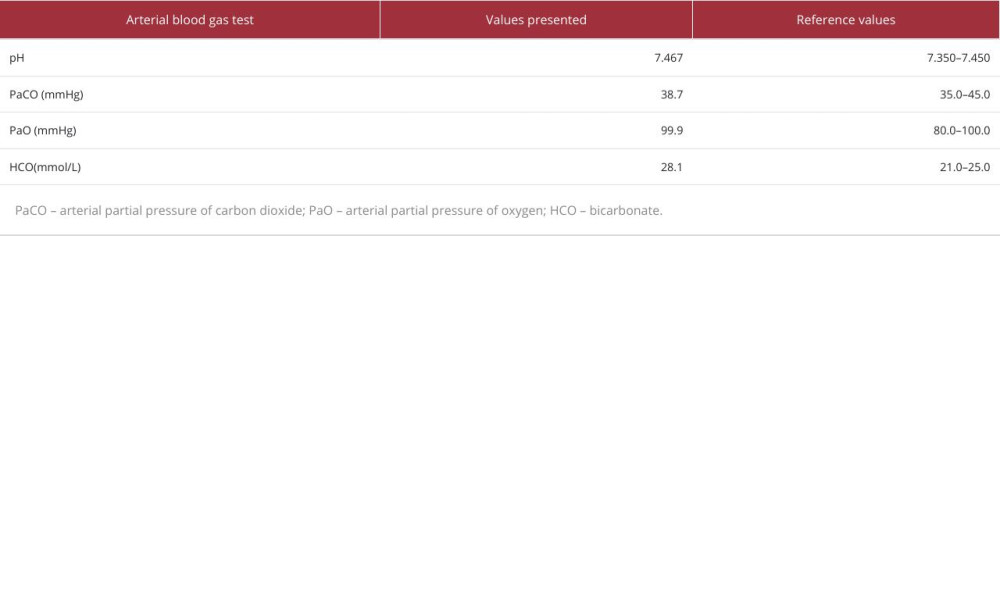 Table 2.. Arterial blood gas test results.
Table 2.. Arterial blood gas test results.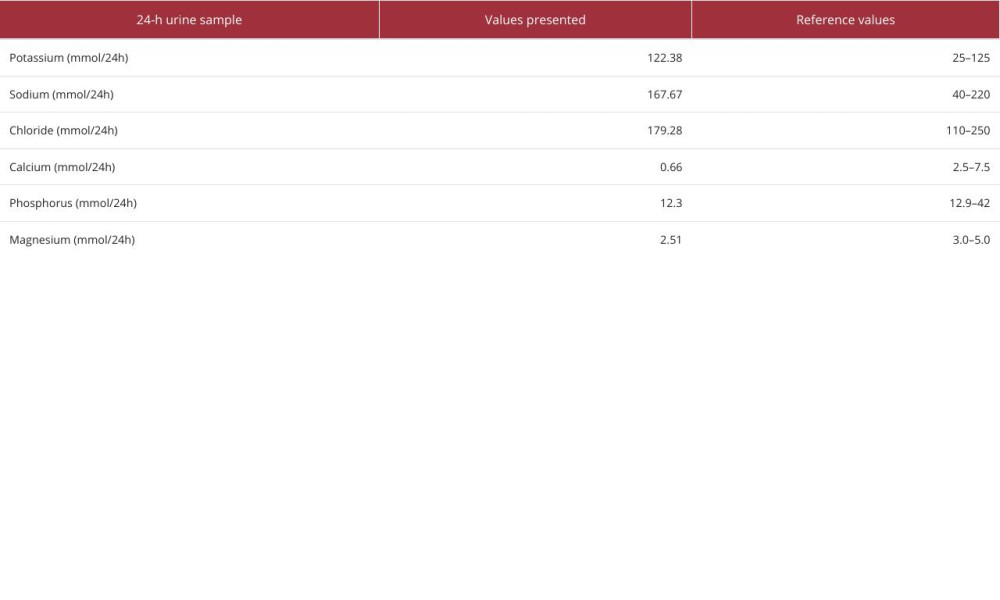 Table 3.. Results of 24-h urine sample biochemical tests.Table 1.. Laboratory findings of the patient on admission.Table 2.. Arterial blood gas test results.Table 3.. Results of 24-h urine sample biochemical tests.
Table 3.. Results of 24-h urine sample biochemical tests.Table 1.. Laboratory findings of the patient on admission.Table 2.. Arterial blood gas test results.Table 3.. Results of 24-h urine sample biochemical tests. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133