04 July 2025: Articles 
Navigating Diagnostic Ambiguities in Cardiac Amyloidosis: Insights from a Case with Delayed Diagnosis of AL Amyloidosis
Unusual clinical course, Challenging differential diagnosis, Unusual or unexpected effect of treatment, Diagnostic / therapeutic accidents, Unexpected drug reaction, Educational Purpose (only if useful for a systematic review or synthesis)
Antonio Al Hazzouri ADEF 1, Rose-Mary DaouDOI: 10.12659/AJCR.948092
Am J Case Rep 2025; 26:e948092






Abstract
BACKGROUND: Cardiac amyloidosis is the accumulation of aberrant proteins in the heart, liver, brain, and several other organs. It presents both extracardiac and cardiac symptoms, making diagnosis difficult and early detection crucial in the prognosis of the patient. Diagnostic techniques for cardiac amyloidosis can present false-negative results, making diagnosis difficult in the early stage of the disease.
CASE REPORT: A 67-year-old man presented for worsening dyspnea of several months’ duration and recent cough. Echocardiography study showed unexplained biventricular hypertrophy with preserved left ventricular systolic function, and a restrictive filling pattern suggesting an infiltrative heart disease like cardiac amyloidosis. Technetium pyrophosphate scan, light-chain assay, and serum and urine protein immunofixation were negative. The patient was treated as a case of advanced heart failure, with initial improvement. He started deteriorating progressively, and repeating the workup was considered. Six months later, a repeated echocardiogram showed severely impaired left ventricular systolic function and findings suggestive of advanced cardiac amyloidosis. Free light-chain assay was positive, in favor for AL amyloidosis, which was confirmed by a bone marrow biopsy and cardiac MRI. Chemotherapy was started, but the patient died due to stage D heart failure caused by advanced AL amyloidosis.
CONCLUSIONS: We aim at increasing awareness of the early diagnosis of cardiac amyloidosis and highlighting the importance of considering the disease even with an initial negative workup. We will also try to explain the reason for the false-negative initial workup and to implement the use of cardiac MRI in early stages if the clinical suspicion for the disease is high.
Keywords: Case Reports, Heart Failure, Echocardiography, Amyloidosis, Humans, Male, Aged, delayed diagnosis, Fatal Outcome, Immunoglobulin Light-chain Amyloidosis, Cardiomyopathies
Introduction
The hallmark of amyloidosis is the abnormally accumulated insoluble polymeric fibrillar proteins outside the cells, usually in blood vessels and organs. Based on the amyloid protein type, cardiac amyloidosis can be classified into 3 variants: (1) AL (immunoglobulin light-chain) amyloidosis, which is the most common type, (2) ATTRwt (“senile cardiac amyloidosis”), and (3) transthyretin amyloid cardiomyopathy (ATTRM) leading to hereditary cardiac amyloidosis due to mutated transthyretin [1]. Several diagnostic strategies are used in cardiac amyloidosis (CA), including electrocardiography, echocardiography, cardiac magnetic resonance imaging (C-MRI), diphosphonate scintigraphy, positron emission tomography, and cardiac biomarkers [2]. Since the disease can affect various organs, a variety of symptoms can present. In addition, isolated cardiac amyloidosis is a rare finding, which makes its diagnosis difficult. Furthermore, the diagnostic techniques of cardiac amyloidosis can produce false-negative results, which can lead to delayed diagnosis of the disease and poorer prognosis. Several reported cases, discussed later, had an initial negative workup, but at least 1 test additional to echocardiography was positive. Thus, cardiac imaging through echocardiography or C-MRI is critical to establish a preliminary diagnosis, and it is important to detect typical findings that are highly specific in diagnosing the disease. One example is the “cherry on top” sign, which consists of impaired longitudinal strain (LS), with the apical segments remaining relatively unaffected, and it has a 100% sensitivity for CA. Concomitant CA and aortic stenosis (AS) can be misdiagnosed as isolated AS, potentially affecting treatment decisions and patient outcomes. Echocardiographic findings showed that the E/E’ ratio and regional apical longitudinal strain (RALS) were significantly higher in CA patients, while global longitudinal strain (GLS) and basal LS were lower [3].
Here, we present the case of a 67-year-old nonsmoking Arab man presenting for dyspnea of several months with sporadic cough and no symptoms of congestive heart failure (CHF). He was known to have prostate cancer, hypothyroidism, and a history of stroke. Transthoracic echocardiography (TTE) was done and was highly suggestive of CA, so further investigations were done, including technetium pyrophosphate scan (PYP), immunoelectrophoresis, urine electrophoresis, and serum and urine immunofixation of proteins, which were all negative for cardiac amyloidosis, in addition to bone marrow biopsy. The patient was then treated as a case of heart failure with preserved ejection fraction (HFpEF) and we repeated the laboratory workup after 6 months. During this time frame, several complications were encountered, including a stroke related to paroxysmal atrial fibrillation, lower-limb edema, pleural effusion, and ascites refractory to diuresis. Echocardiography was repeated and findings were again suggestive of cardiac amyloidosis. Blood workup was also repeated and showed AL amyloidosis, confirmed on C-MRI, but the patient died due to stage D heart failure, although he was adherent to and following on his treatment and interventions through regular visits and follow-ups with his treating physician. The aim of our case report is to show that various diagnostic tools are applicable in the diagnosis of CA, with echocardiography as the first-line tool that can provide clues about the presence of CA, but blood workup findings are sometimes negative and do not always support the presence of CA until after the disease progresses.
Case Report
A nonsmoking 67-year-old man with no known food or drug allergies presented for dyspnea of several months’ duration with recent sporadic dry cough, no angina-like symptoms, no CHF-LIKE symptoms, and no orthopnea or paroxysmal nocturnal dyspnea. His dyspnea began a few months before presentation, when he visited a pulmonologist who proposed that the cough was allergic in nature and prescribed symptomatic treatment. He then saw another physician after no resolution of symptoms, who ordered a chest CT scan. The scan showed moderate bilateral pleural effusion, aortic calcification with an aorta of 4.2 cm diameter, and left lower-lobe retro-cardiac atelectasis. His past medical history and past surgical history included prostate cancer s/p transurethral resection of the prostate (2016), adenoidectomy (2016), hypothyroidism, and an old stroke of unknown etiology as per the patient. His family history was significant for prostate cancer. A laboratory workup showed a negative troponin level and a N-terminal prohormone brain natriuretic peptide (NT-proBNP) level of 525 ng/L. Initial TTE showed restrictive filling pattern with an E/E’: 19.88, MV E’A of 2.8, MV E velocity of 0.67m/s (Figure 1), unexplained right (Figure 2) and left ventricular hypertrophy with preserved left ventricular (LV) systolic function, speckled appearance of myocardial tissue (Figure 3), severe tricuspid regurgitation (Figure 4), bi-atrial enlargement and trace pericardial effusion and significant pleural effusion (Figure 5). These findings were highly suggestive of cardiac amyloidosis. Therefore, a technetium pyrophosphate scan, ultrasound (U/S) abdomen, free light-chain assay, and serum/urine immunofixation of proteins were ordered to determine the type of amyloidosis. PYP was negative for ATTR (grade 0). Immunoelectrophoresis was negative: IgG 5.04, IgA: 2.15, IgM: 0.383, kappa: 1.28, lambda: 0.89. Urine electrophoresis was also negative: free kappa: 140, free lambda: 132, immunofixation of proteins showed absence of biochemical signs of monoclonal gammopathy, and urine immunofixation showed absence of Bence Jones proteins. Also, fat pad biopsy and bone marrow were negative, ruling out AL cardiac amyloidosis. The patient was treated as a case of HFpEF with bisoprolol, bumetanide, and empagliflozin 10 mg and planned to have a follow-up TTE after 6 months. During this time frame, the patient had a stroke related to paroxysmal atrial fibrillation and clinical deterioration characterized by borderline blood pressure, lower-limb edema, pleural effusion, and ascites, which were refractory to oral diuresis. The patient had been adherent and following up regularly on his treatment and interventions with his treating physician. Several hospital admissions were needed for intravenous diuresis. Due to the worsening of his clinical status, repeating the whole workup was planned. Repeated TTE showed overall LV systolic function that is severely impaired with an EF of 20–25%, a very low septal E’ (Figure 6), and severe diastolic dysfunction (Grade III) with right-ventricle (RV) hypertrophy measuring 10 mm. The left atrium was severely dilated, with severe spontaneous echocardiography contrast, and the inferior vena cava was dilated, with poor inspiratory collapse. This constellation of findings was again suggestive of CA. The blood workup was repeated and the results suggested AL amyloidosis, with a poor prognosis. These blood test results showed an increase in plasma free light-chain and a shift in kappa/lambda ratio, suggesting AL amyloidosis progression. C-MRI showed diffuse transmural late gadolinium enhancement (LGE) and abnormal myocardial nulling pattern suggesting advanced CA with biventricular failure. The patient died due to stage D heart failure caused by advanced AL amyloidosis after a very late and short trial of chemotherapy. A summary of the clinical progression of the patient along with the corresponding workup and management is provided in Table 1.
Discussion
The classification of CA is determined by the type of amyloid protein involved. The AL variant, also known as primary amyloidosis, is the most common. The senile amyloidosis (SA) variant is the least common type that has cardiac presentations [1]. Accumulation of light immunoglobulin chains due to plasma-cell dyscrasia causes AL amyloidosis [1]. Hereditary ATTR can have both cardiac and neurological presentations. People diagnosed with ATTRwt tend to have more arrhythmias than individuals possessing hATTR. All types of amyloidosis lead to restrictive cardiomyopathy with preserved diastolic dysfunction presenting as HFpEF [1].
Symptoms associated with CA are mostly related to right-heart failure, and include typical or atypical angina, orthopnea, paroxysmal nocturnal dyspnea, fatigue, pre-syncope, and syncope [1]. Other features include peripheral edema, ascites hepatosplenomegaly, and pleural effusion can be present [4,5]. Atrial rhythm disturbances are common, increasing the risk of atrial fibrillation (AF) and thus embolic phenomenon [1,6]. Extracardiac signs usually precede cardiac manifestations in most individuals. In AL amyloidosis, the major findings are macroglossia, periorbital purpura, and nail dystrophy [1]. Senile and hereditary amyloidosis can cause a neuropathy-dominated picture, as well as carpal tunnel syndrome, tendon rupture, and an ascending symmetrical length-dependent sensorimotor axonal polyneuropathy [1,6].
The diagnosis for CA includes both invasive and non-invasive methods. The invasive criteria can be used in all forms of cardiac amyloidosis, whereas the non-invasive one is applied only for ATTR [7]. Chronically elevated troponin in the presence of left ventricular hypertrophy (LVH) and in the absence of other explanation should prompt a workup for amyloidosis. Additional markers, including glomerular filtration, are also part of the backbone of prognostic models in both AL-CA and ATTR-CM [8].
Current guidelines employ this algorithm (Figure 7) to approach CA and differentiate between their types. However, in our case this algorithm did not reach the diagnosis of cardiac AL amyloidosis until the second set of lab tests were ordered several months later. Initially, blood tests for monoclonal proteins were negative as well as bone scintigraphy. Later, the blood tests were positive and cardiac MRI confirmed our suspicion.
In individuals with suspected CA, the most commonly used technique of imaging and assessment is echocardiography (Figure 7) [9]. The most common finding detected by echocardiography is left ventricular hypertrophy (LVH) leading to decreased ventricular size, which is symmetrical in AL-CA patients but asymmetrical in ATTR-CA patients. Other findings include bi-atrial enlargement, heart failure with reduced ejection fraction in late stage, restrictive diastolic dysfunction, myocardial echogenicity, and pericardial effusion. HFpEF and restrictive cardiomyopathy with diastolic dysfunction are other findings in patients with early-stage cardiac amyloidosis. Thrombosis is a major consequence of left atrial (LA) enlargement in individuals with cardiac amyloidosis due to atrial remodeling, endocardial disturbances, hypercoagulable state, and diastolic dysfunction [9].
In our case, initial echocardiography showed a speckled myocardial appearance, mild LVH with fair LV systolic function with an EF of 50–55% and severe diastolic dysfunction (Grade III) with elevated filling pressure, bi-atrial enlargement, and trace pericardial effusion. The RV was moderately hypertrophied, measuring 10 mm. Six months later, repeated echocardiography showed mild concentric LVH and severely impaired LV systolic function, with EF estimated at 20–25% and trivial pericardial effusion.
Serum and urine immunofixation electrophoresis and serum-free light-chain (sFLC) are considered as initial evaluations for suspected AL amyloidosis after initial echocardiography (Figure 7) [10]. The presence of an M-spike on electrophoresis indicates possible monoclonal protein. However, complementation by immunofixation of the serum and urine and calculation of sFLC (ratio of free kappa over lambda light-chain levels) is recommended [11]. Studies have shown that 30% of patients with CA have normal serum and urine protein electrophoresis with abnormal immunofixation [10]. An abnormal kappa-lambda ratio is detected by the sFLC assay in patients with AL amyloidosis. The hallmark of AL amyloidosis is the high frequency of lambda light-chain proteinemia. If Ig FLC (κ: λ) ratio is normal (0.26–1.65) and immunofixation of serum and urine is negative, then AL amyloidosis is unlikely and further workup should be done, unless clinical suspicion is high [4]. A retrospective study on 98 patients with AL amyloidosis confirmed by biopsy studied the detection of light-chain clonality among different techniques. In descending order of sensitivity, ferric carbosxymaltose (FCM) assay identified aberrant BM plasma cells in 92.9% of cases, bone marrow (BM) immunohistochemistry (IHC) identified 85.7%, sFLC ratio identified 79.5%, serum immunofixation electrophoresis (IFE) identified 63.3%, urine IFE identified 70.8% [12], and IHC for light chains in BM biopsies usually reveals monoclonal plasma cells. However, in cases with small numbers of clonal plasma cells, they may be masked by normal polyclonal plasma cells [12]. Immunoelectrophoresis in our case was negative, as well as urine electrophoresis. Immunofixation of proteins showed absence of biochemical signs of monoclonal gammopathy of undetermined significance and urine immunofixation showed the absence of Bence Jones proteins.
There are several indications for biopsy in ATTR-CM: (1) when it is hard to reliably differentiate between AL and ATTR-CM, such as in an abnormal monoclonal protein workup in elderly individuals who are at risk for ATTR-CM or in conjunction with abnormal bone scintigraphy (Figure 8); (2) when there are atypical findings on imaging, such as borderline or negative nuclear scintigraphy in patients with suspected ATTR-CM; and (3) to evaluate for other less common forms of amyloidosis [8]. The diagnosis of CA is confirmed when an endomyocardial biopsy with Congo red staining demonstrates amyloid deposits irrespective of the degree of LV wall thickness. Mass spectrometry remains the preferred technique for determining the type of amyloid [7]. The sensitivity of Congo red staining for AL amyloidosis differs significantly based on specimen source – 69% for bone marrow biopsy, 75% for fat pad aspiration, and 100% for heart biopsy. Thus, negative results of peripheral biopsy do not rule out an AL diagnosis [13]. Although immunostaining is the technique most commonly used by surgical pathologists for subtyping amyloid, it has many problems [14], including false-negative results due to weak or no staining (commonly seen in commercial antibodies to kappa and lambda light chains 11 and 16 due to presence of truncated light chains in AL type amyloid deposits) and false-positive results due to non-specific immunostaining of the amyloid deposits [14]. In a study performed in 1964, Congo red staining showed a 24.3% false-negative rate and a 4.2% false-positive rate, and the chief cause of false-negative results was the minimal presence of amyloid [12]. Another reason for a false-negative result is minimal myocardial amyloid deposition in the early stages of the disease. Cardiac amyloidosis has several types, including ATTRwt, formerly known as senile cardiac amyloidosis, which develops as amyloid fibrils gradually accumulate in the myocardium over many years. In the initial phases, the amyloid level may be too low for detection. Another cause of false-negative results is related to prior myocardial infarction, but for a different reason than false-positive findings – an old (remote) myocardial infarction leads to the formation of scar tissue, which lacks perfusion and does not accumulate amyloid fibrils [15].
The results obtained in our case regarding the biopsy with Congo red stain and immunostaining for AAC were negative, but this does not exclude the presence of amyloid deposition or plasma cell dyscrasia. The sensitivity of Congo red staining in bone marrow biopsy for AL amyloidosis is only about 69%, and minimal amyloid presence or a low clonal plasma cell burden can result in false negatives. As noted earlier, small numbers of clonal plasma cells may be masked by polyclonal plasma cells, leading to missed diagnoses. Therefore, a negative biopsy does not definitively rule out AL amyloidosis, particularly in the early stages of the disease.
In the absence of histology, cardiac ATTR amyloidosis can be diagnosed in the setting of typical echocardiographic/CMR findings when 99mTc-PYP shows grade 2 or 3 myocardial uptake of radiotracer, and can also be diagnosed based on sFLC assay and by serum and urine protein electrophoresis with immunofixation to exclude clonal dyscrasia (Figure 7) [7]. When the disease is suspected, the specificity of Grade 2 or 3 bone scintigraphy for cardiac ATTR has been proposed to be almost 100% in the absence of a detectable monoclonal protein and an abnormal serum FLC ratio. Then, we proceed with genetic testing to differentiate between ATTRv and ATTRwt forms. No cardiac uptake on scintigraphy and negative monoclonal protein assessments indicate a very low probability of CA [7]. In our case, the PYP was negative for TTR amyloidosis (grade 0).
Staging cardiac amyloidosis can help predict patient prognosis and response to chemotherapy. The European modification of the Mayo 2004 staging system classifies AL amyloidosis patients based on troponin T (TnT) >0.035 mcg/L and NT-proBNP >332 ng/L into 3 stages: Stage I (normal levels), Stage II (1 elevated biomarker), and Stage III (both elevated), with median survival times of 26, 11, and 4 months, respectively. The European revision further splits Stage III into IIIA (NT-proBNP ≤8500 ng/L) and IIIB (NT-proBNP >8500 ng/L). The Mayo 2012 staging system introduced dFLC >18 mg/dL and adjusted cardiac biomarker thresholds (NT-proBNP >1800 ng/L, TnT >0.025 mcg/L), expanding the classification to 4 stages based on the number of elevated risk factors, with zero risk factors constituting Stage I. Median survival times were 94, 40, 14, and 6 months for Stages I–IV, respectively, with improved outcomes in HCT-eligible patients (4-year survival: Stage I, not reached; Stage II, 97 months; Stage III, 58 months; and Stage IV, 22 months). AL amyloidosis has a poor long-term prognosis if detected at a late stage of the disease, with a median survival as short as 5 months [16]. In 2016, Grogan et al developed the first prognostic score for ATTR-CA in a cohort of patients with ATTRwt-CA. This score utilized NT-proBNP with a threshold of 3000 ng/L and TnT with a cutoff of 50 ng/L to classify patients into 3 categories based on increasing mortality risk [17]. A study done by Oubari et al (2021) found a correlation between the severity of cardiac disease, as classified by the modified Mayo 2004 stages, and hematologic response to first-line treatment. Patients in Mayo stages I, II, and IIIa achieved complete remission (CR) or very good partial remission (VGPR) in 67%, 58%, and 63% of cases, respectively. However, those in Mayo stage IIIb had a significantly lower response rate, with only 40% achieving CR or VGPR, highlighting a strong association between advanced cardiac involvement (Stage IIIb) and poorer hematologic response [18].
Used as an adjunct to echocardiography, CMR can characterize myocardial tissue, which gives it a diagnostic advantage over echocardiography. It can differentiate CA from non-amyloid wall-thickening cardio myopathies and has a unique strength in determining disease prognosis and treatment response. However, it is not the first-line modality due to the expertise required, cost, lack of widespread use, and limitation with renal impairment [19].
LGE, is the main tool used in CMR to assess the presence of CA, but it has some limitations. First, it is a qualitative marker and cannot quantify amyloid burden. Second, it is contraindicated in patients with severe renal impairment with a glomerular filtration rate (GFR) <30 ml/min. In addition, some patients with CA do not exhibit LGE on CMR, especially when patients are scanned too early in the disease [20]. A recent meta-analysis of 7 studies found that CMR-based LGE had a sensitivity of 85% and specificity of 92% for diagnosing cardiac amyloidosis. These results emphasize LGE’s high sensitivity as a diagnostic marker for cardiac amyloidosis, but also indicate that LGE may not be present in the early stages of the disease [21]. A relatively novel technique in diagnosis and prognostication of CA is quantification of myocardial extracellular volume (ECV) by combining contrast-enhanced T1 with native T1. ECV is a marker of myocardial tissue remodeling similar to T1, but T1 incorporates both extracellular and intracellular factors, which are strongly affected by water content, such as in edema. Therefore, ECV, which is elevated in CA patients compared to healthy and other pathologies, is superior in providing a true quantification of amyloid burden and has high sensitivity (92%) and specificity (82%) for diagnosing ATTR [19,22].
Patients with an ECV of 0.30% or less, indicating no cardiac amyloid infiltration, had significantly better survival rates compared to those with higher ECV. Mortality increased with greater degrees of cardiac involvement.
For patients with ECV ≤0.30%, long-term survival was unaffected by hematological response. In those with ECV between 0.31% and 0.40% (early AL amyloidosis), non-responders (NR) had poorer survival. Patients with ECV between 0.41% and 0.50% had better survival when achieving CR or VGPR, while partial responders (PR) and non-responders had worse outcomes. When ECV exceeded 0.50%, survival was favorable only with CR, while VGPR, PR, and NR were associated with worse outcomes.
ECV mapping has emerged as the strongest independent predictor of prognosis, offering insights into the necessary depth and speed of hematological response. Not all patients required a rapid response, but those with higher cardiac infiltration benefited from early and deep hematological response. These findings suggest that adjusting treatment based on pretreatment ECV and hematological response could improve survival outcomes [23].
The concept behind CA treatment relies on supportive care, which aims to alleviate symptoms associated with arrhythmias, conduction abnormalities, and heart failure. Along with drugs like aldosterone antagonists and loop diuretics, lifestyle changes like dietary adjustments and sodium restriction are advised for heart failure (Figure 8). Due to their high affinity for fibrils and negative inotropic effect, calcium channel blockers (CCBs) cannot be administered. Anticoagulation is advised in cases of AF according to guidelines and consensus, regardless of the CHA2DS2-VASc risk score, with direct oral anticoagulants approved as first-line treatment (Figure 8). Heart resynchronization therapy and permanent pacemaker implantation are the most recommended treatments for conduction dysfunction in CA. In addition, specific amyloidosis therapy is recommended. For patients with AL amyloidosis, chemotherapy followed by stem cell transplantation is the required treatment (Figure 8). Proteasome inhibitors are also occasionally used [1]. Disease-modifying treatments used in CA include TTR disrupters (eg, doxycycline, tauro ursodeoxycholic acid), TTR stabilizers (eg, tafamidis, diflunisal), and TTR silencers (eg, patisiran, inotersen, vutrisiran) (Figure 8) [24,25]. Plasma cells are the newest modalities targeting monoclonal antibodies. Daratumumab is a human monoclonal antibody that targets cluster of differentiation (CD38), which is highly expressed in aberrant plasma cells. In early trials, daratumumab showed encouraging results for patients with AL amyloidosis [1]. In a retrospective trial, the hematological response was obtained in 76% of cases (36% with complete remission) [25].
There are atypical cases of cardiac amyloidosis with negative workup. One case had negative results for serum and urine immunofixation, a normal serum kappa/lambda light-chain ratio, and negative results of genetic testing of the
In our case, unlike typical cases of AL cardiac amyloidosis, the initial blood tests did not detect early-stage disease following the standard diagnostic protocol. Subsequently, with disease progression, blood tests showed slight positivity, prompting the use of cardiac MRI to confirm the suspicion of amyloidosis based on echocardiography findings. Laboratory workups and biopsies can produce false-negative results. Therefore, relying on an initial negative workup can be misleading and lead to a late diagnosis of the disease and poor prognosis. C-MRI was found to have a high sensitivity, and we believe that it should have been implemented earlier, but unfortunately it was not available at that time. We considered the blood workup to be specific based on the low burden of disease and the few amyloid deposits present in the early stage of the disease, and these indicators were expected to rise over time and considering our patient’s symptoms had been present for several months. However, this was not the situation with our patient, and this strengthens the recommendation for early C-MRI. Having diagnosed the disease earlier, the patient would have started chemotherapy early in the disease, as he had a low Mayo staging, expecting a good response to therapy. This shows that the standard diagnostic approach can miss typical presentations of cardiac amyloidosis, emphasizing the need for further investigation when the diagnosis is strongly suspected and recommending early use of C-MRI in highly suspicious cases.
Conclusions
In summary, cardiac amyloidosis is a disease with various pathogeneses and clinical features on presentation that can be manifested by cardiac and extracardiac symptoms, making it challenging to diagnose. Various diagnostic techniques are applicable, each having its own advantages and limitations; however, as in our case, it is sometimes difficult to diagnose a patient with cardiac amyloidosis, as the diagnostic workup can lead to false-negative results. Thus, more sensitive and specific diagnostic tools are required for rapid detection of the disease, like cardiac MRI, to help implement early treatment and prevent progression of the disease, as in our case the patient was initially classified as Stage II based on the Mayo staging and would benefit from early chemotherapy. Future diagnostic approaches should emphasize the integration of early C-MRI and serial free light-chain assays in patients with unexplained restrictive cardiomyopathy. Our rare and unique case report aims to increase awareness of the early diagnosis of cardiac amyloidosis despite its varied clinical manifestations, thereby providing scientific and educational merit.
Figures
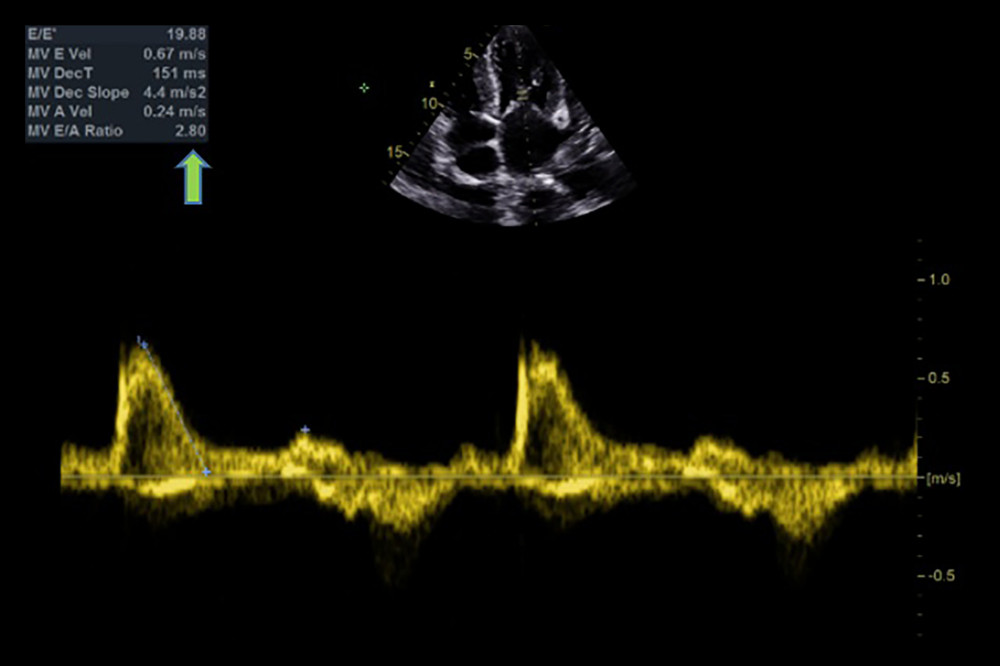 Figure 1. Echocardiographic findings showing restrictive filling pattern with E/E’: 19.88 and an E/A ratio >2 (arrow), a hallmark of cardiac amyloidosis.
Figure 1. Echocardiographic findings showing restrictive filling pattern with E/E’: 19.88 and an E/A ratio >2 (arrow), a hallmark of cardiac amyloidosis. 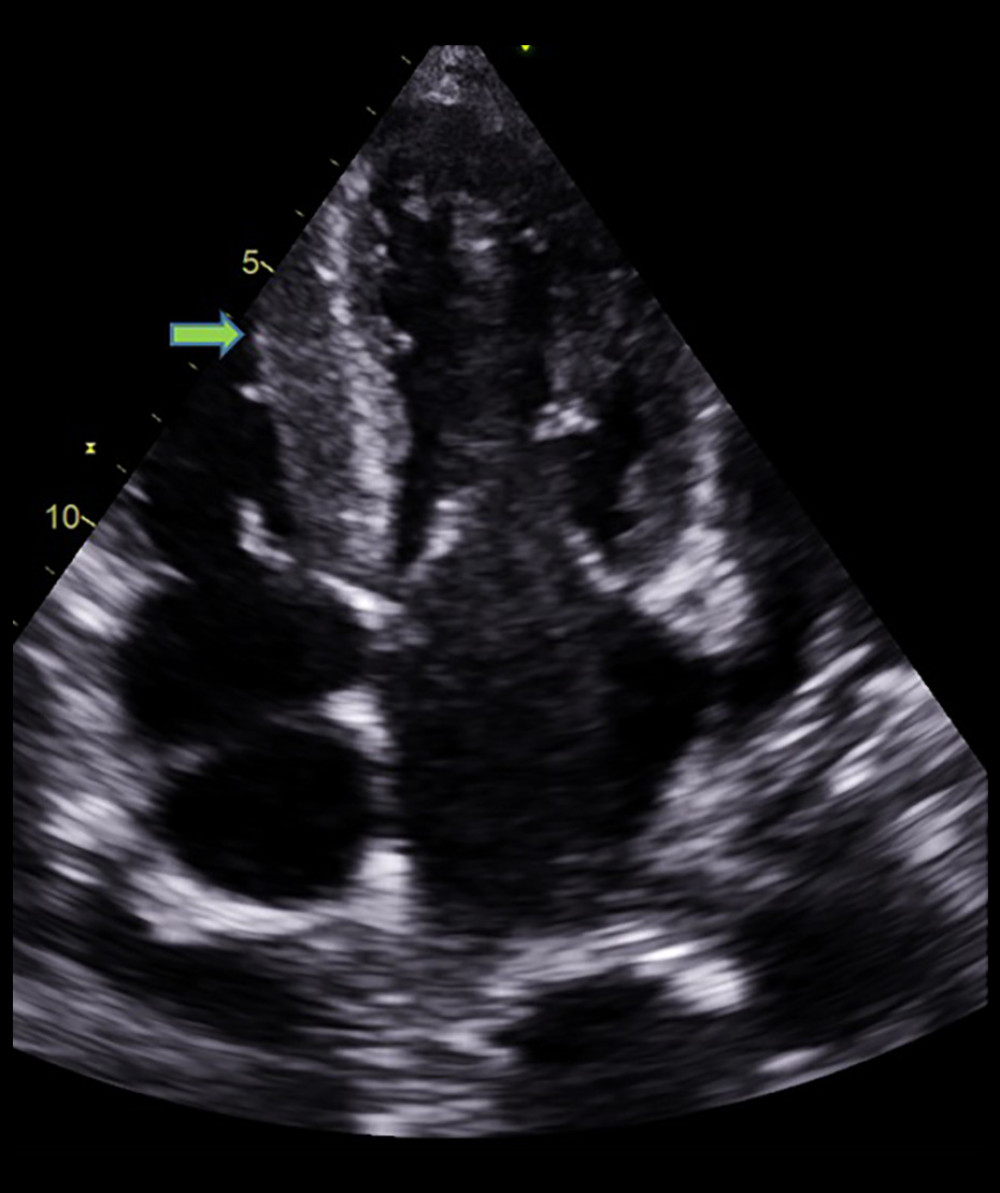 Figure 2. Echocardiographic finding showing right ventricle hypertrophy (arrow) and spontaneous echo contrast in left atrium.
Figure 2. Echocardiographic finding showing right ventricle hypertrophy (arrow) and spontaneous echo contrast in left atrium. 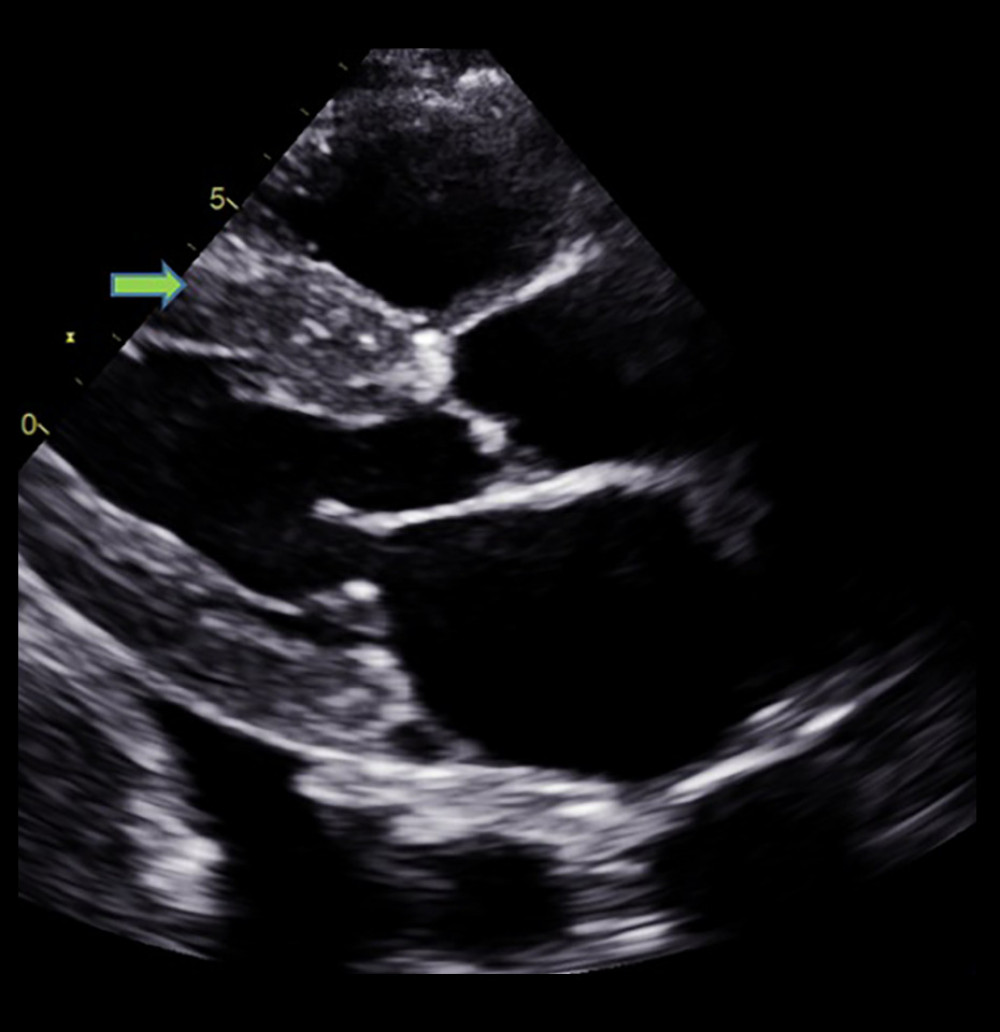 Figure 3. Echocardiographic finding showing left ventricular hypertrophy (arrow) with speckled myocardial appearance and pleural effusion.
Figure 3. Echocardiographic finding showing left ventricular hypertrophy (arrow) with speckled myocardial appearance and pleural effusion. 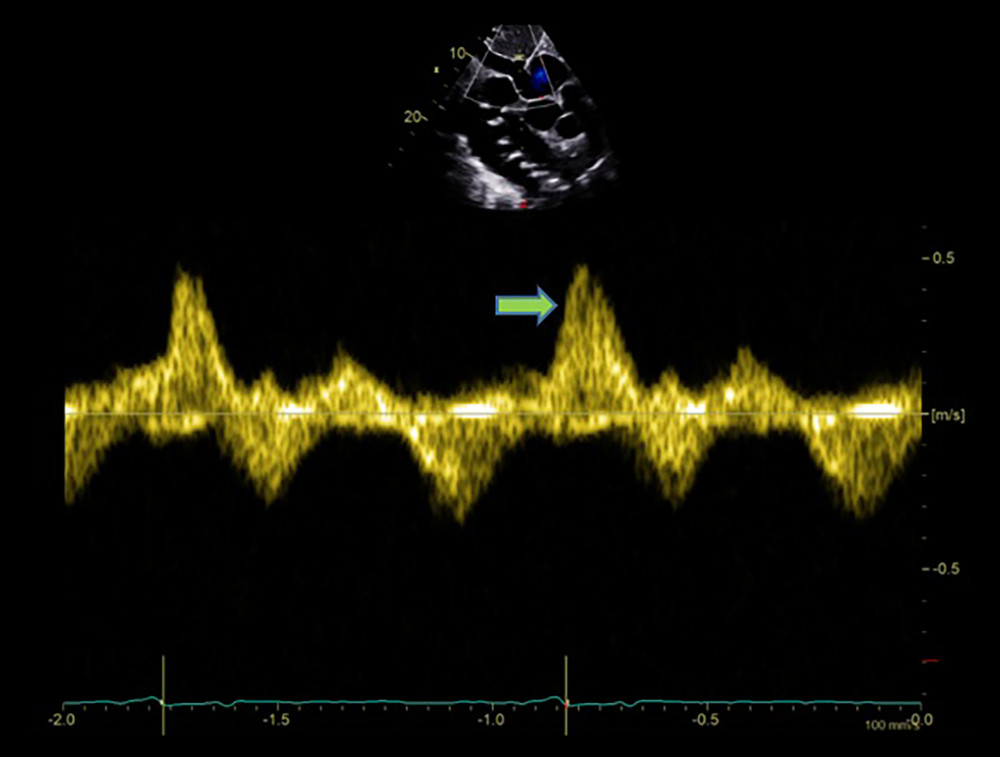 Figure 4. Echocardiographic finding showing systolic flow reversal (arrow) in the hepatic vein indicative of severe tricuspid regurgitation.
Figure 4. Echocardiographic finding showing systolic flow reversal (arrow) in the hepatic vein indicative of severe tricuspid regurgitation. 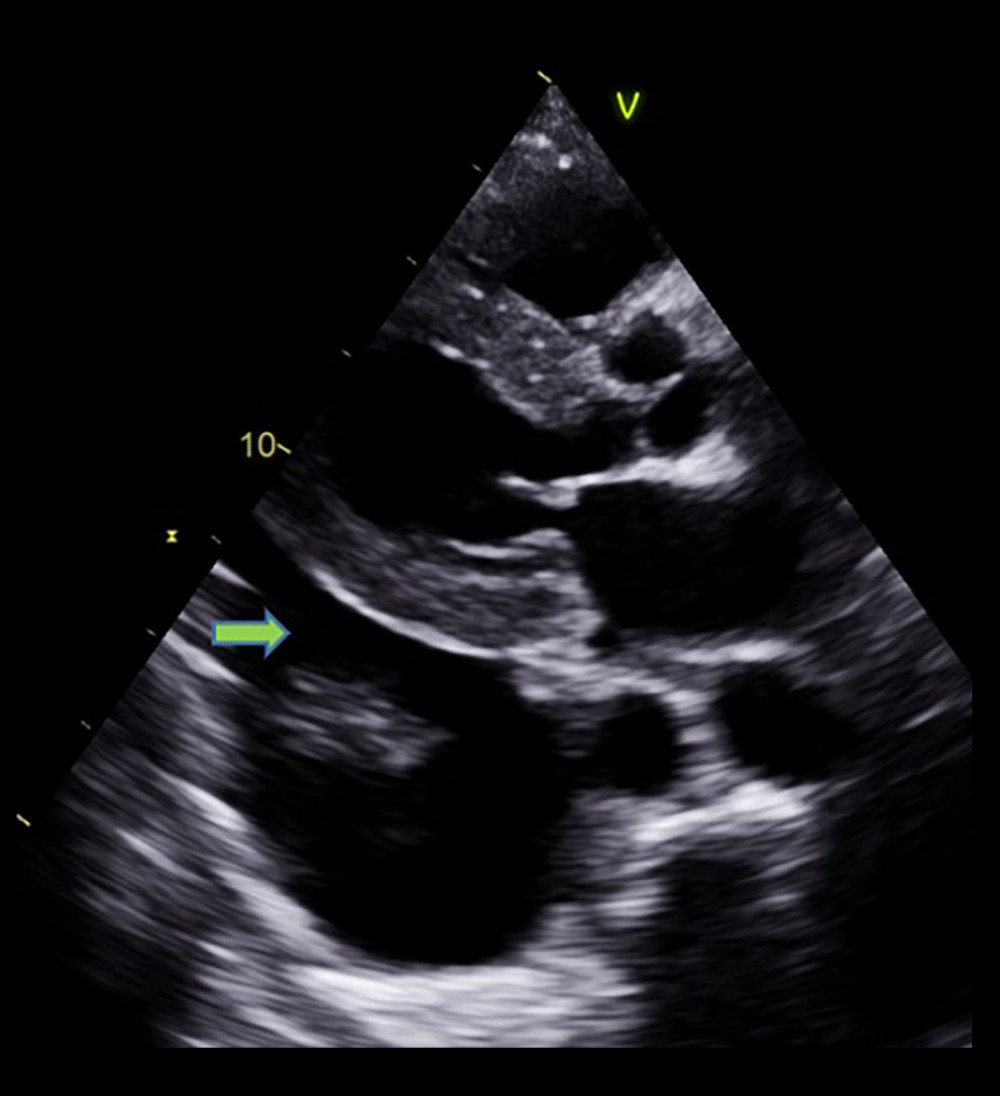 Figure 5. Mild pericardial effusion and significant pleural effusion (arrow).
Figure 5. Mild pericardial effusion and significant pleural effusion (arrow). 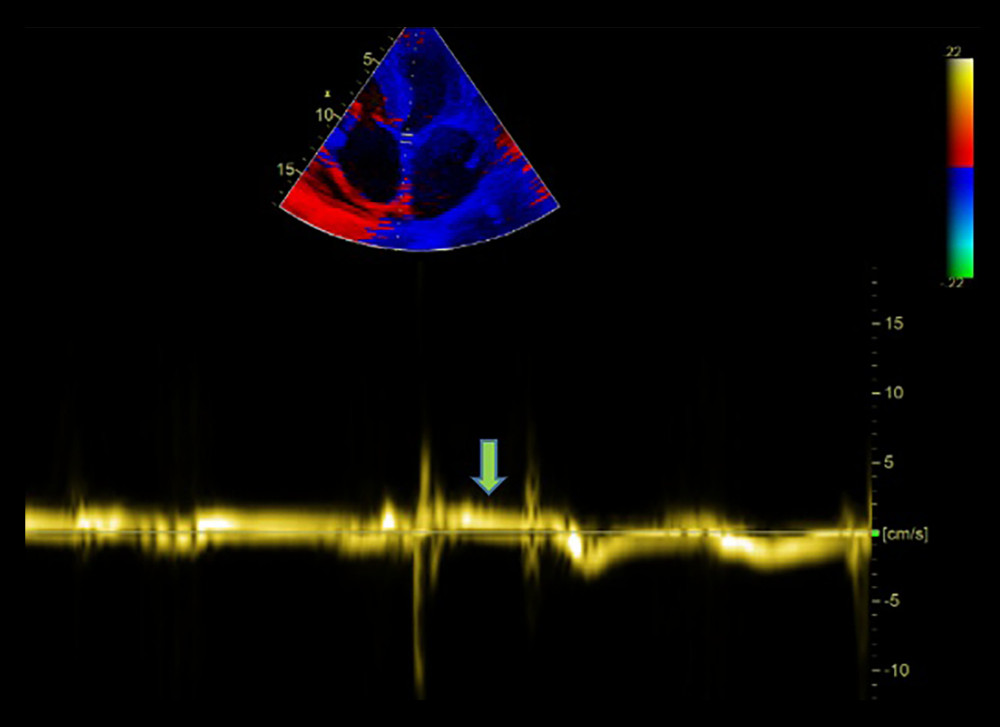 Figure 6. Very low septal E’ (arrow).
Figure 6. Very low septal E’ (arrow). 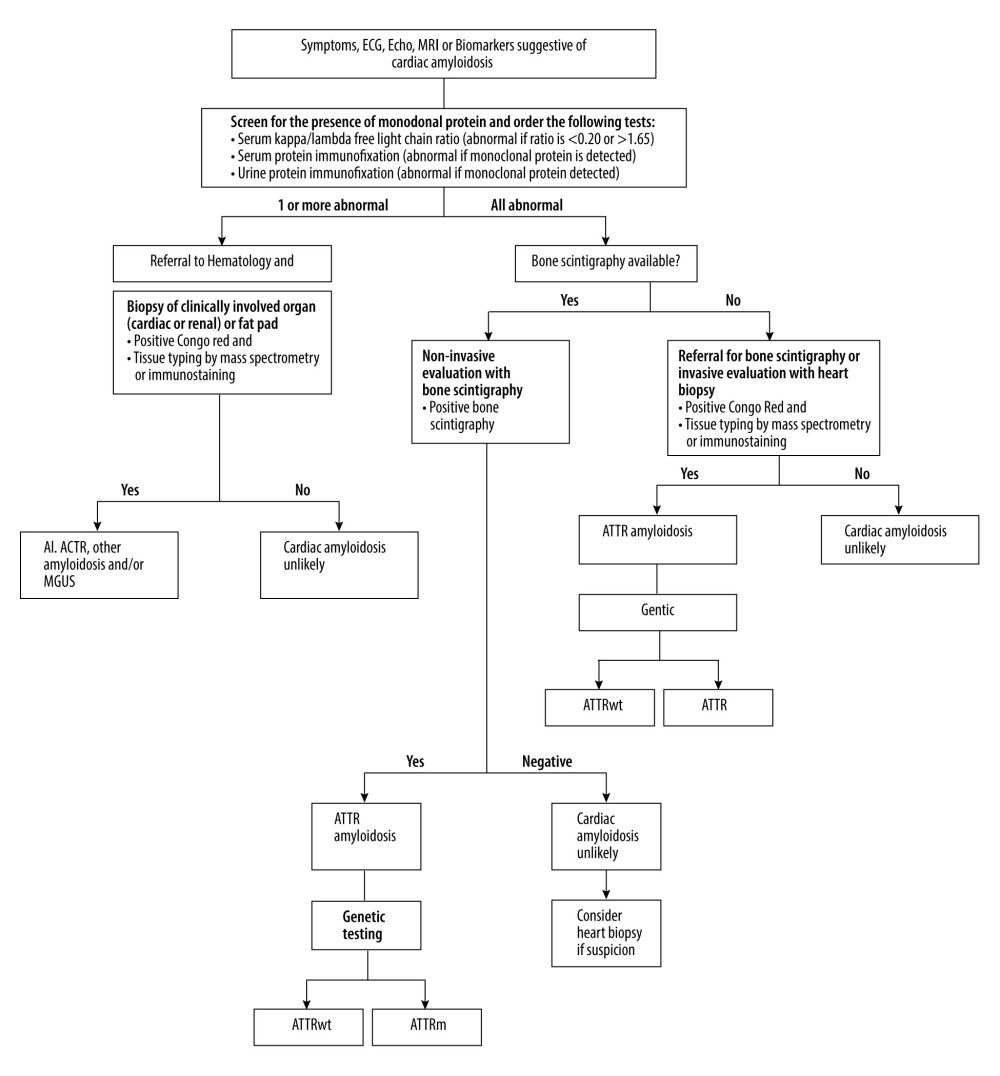 Figure 7. Diagnostic algorithmic approach for cardiac amyloidosis.
Figure 7. Diagnostic algorithmic approach for cardiac amyloidosis. 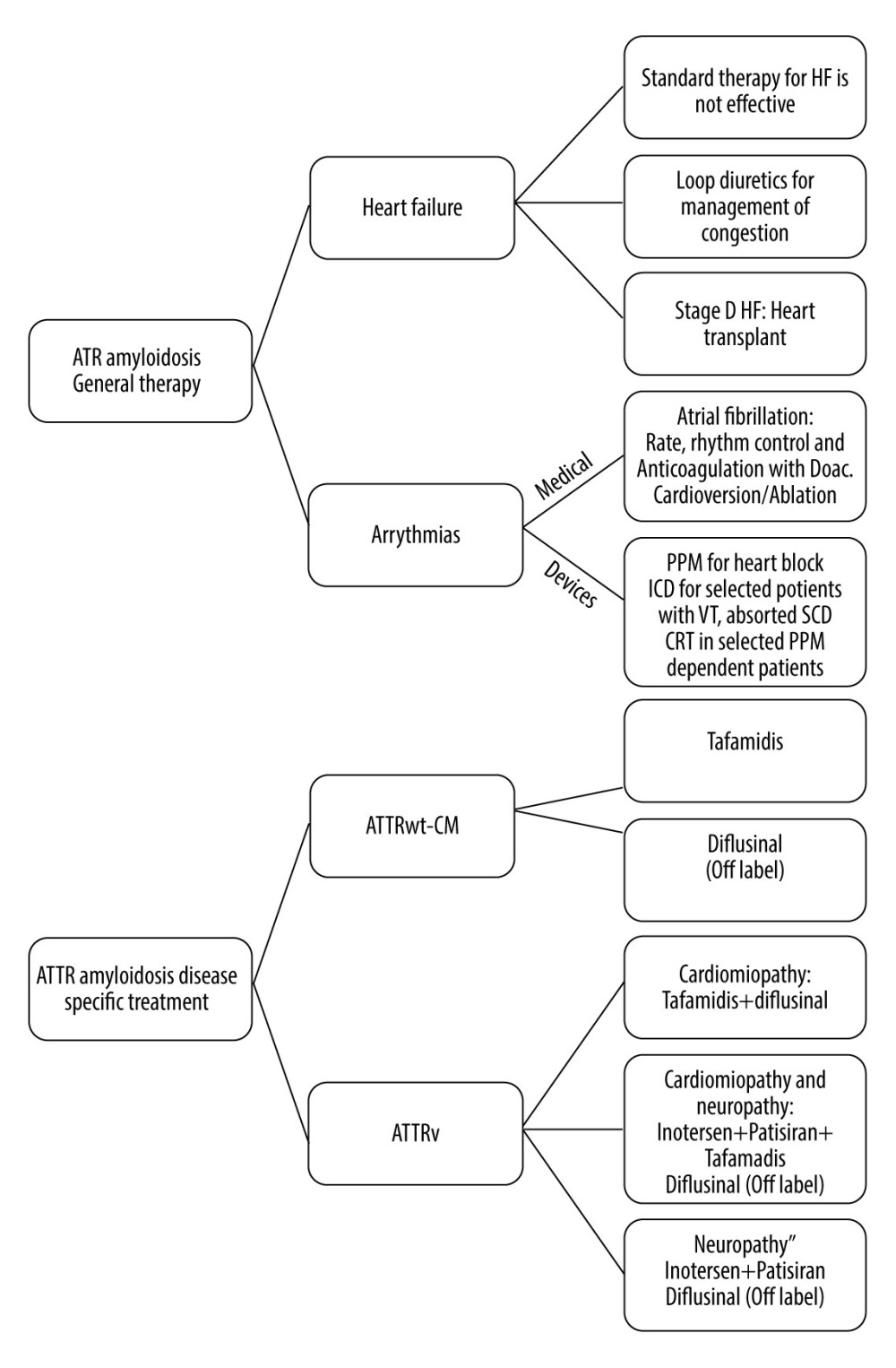 Figure 8. Algorithms that summarize the approach used in treatment of cardiac amyloidosis. Transthyretin amyloidosis (ATTR); Heart failure (HF); Direct oral anticoagulant (DOAC); Cardiac variant transthyretin amyloidosis ATTRwt-CM; Variant transthyretin amyloidosis (ATTRv); Permanent pacemaker (PPM); Implantable cardioverter-defibrillator (ICD); Cardiac resynchronization therapy (CRT); Ventricular tachycardia (VT); Sudden cardiac death (SCD).
Figure 8. Algorithms that summarize the approach used in treatment of cardiac amyloidosis. Transthyretin amyloidosis (ATTR); Heart failure (HF); Direct oral anticoagulant (DOAC); Cardiac variant transthyretin amyloidosis ATTRwt-CM; Variant transthyretin amyloidosis (ATTRv); Permanent pacemaker (PPM); Implantable cardioverter-defibrillator (ICD); Cardiac resynchronization therapy (CRT); Ventricular tachycardia (VT); Sudden cardiac death (SCD). 
References
1. Medarametla GD, Kahlon RS, Mahitha L, Cardiac amyloidosis: Evolving pathogenesis, multimodal diagnostics, and principles of treatment: EXCLI J, 2023; 22; 781-808
2. Brito D, Albrecht FC, de Arenaza DP, World Heart Federation Consensus on Transthyretin Amyloidosis Cardiomyopathy (ATTR-CM): Glob Heart, 2023; 18; 59
3. Jafarisis S, Masoumi S, Khezerlouy-Aghdam N, Strain echocardiography predictors in patients with concomitant cardiac amyloidosis and aortic stenosis: A cross-sectional study: BMC Cardiovasc Disord, 2024; 24; 734
4. Gertz MA, Immunoglobulin light chain amyloidosis: 2022 update on diagnosis, prognosis, and treatment: Am J Hematol, 2022; 97; 818-29
5. Donnelly JP, Hanna M, Cardiac amyloidosis: An update on diagnosis and treatment: Cleve Clin J Med, 2017; 84; 12-26
6. Nakagawa M, Sekijima Y, Yazaki M, Carpal tunnel syndrome: A common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis: Amyloid, 2016; 23; 58-63
7. Garcia-Pavia P, Rapezzi C, Adler Y, Diagnosis and treatment of cardiac amyloidosis: A position statement of the ESC Working Group on Myocardial and Pericardial Diseases: Eur Heart J, 2021; 42; 1554-68
8. Gill SS, Fellin E, Stampke L, Clinical clues and diagnostic workup of cardiac amyloidosis: Methodist DeBakey Cardiovasc J, 2022; 18; 36-46
9. Liang S, Liu Z, Li Q, Advance of echocardiography in cardiac amyloidosis: Heart Fail Rev, 2023; 28; 1345-56
10. Stern LK, Kittleson MM, Updates in cardiac amyloidosis diagnosis and treatment: Curr Oncol Rep, 2021; 23; 47
11. Nijst P, Tang WW, Recent advances in the diagnosis and management of amyloid cardiomyopathy: Fac Rev, 2021; 10; 31
12. Lee T, Park C-J, Kim M, Evaluation of laboratory diagnostic tests for light-chain clonality and bone marrow findings in AL amyloidosis: Blood Res, 2023; 58; 71-76
13. De Michieli L, Light-chain cardiac amyloidosis for the non-expert: Pearls and pitfalls: Intern Emerg Med, 2023; 18; 1879-86
14. Satoskar AA, Efebera Y, Hasan A, Strong transthyretin immunostaining: Potential pitfall in cardiac amyloid typing: Am J Surg Pathol, 2011; 35; 1685-90
15. Jerome S, Farrell MB, Warren J, Cardiac amyloidosis imaging, Part 3: Interpretation, diagnosis, and treatment: J Nucl Med Technol, 2023; 51; 102-16
16. Baker KR, Light chain amyloidosis: Epidemiology, staging, and prognostication: Methodist DeBakey Cardiovasc J, 2022; 18; 27-35
17. Lalario A, Saro R, Sinagra G, Clinical use of biomarkers in cardiac amyloidosis: Heart Fail Clin, 2024; 20; 283-94
18. Oubari S, Naser E, Papathanasiou M, Impact of time to diagnosis on Mayo stages, treatment outcome, and survival in patients with AL amyloidosis and cardiac involvement: Eur J Haematol, 2021; 107; 449-57
19. Bashir Z, Musharraf M, Azam R, Bukhari S, Imaging modalities in cardiac amyloidosis: Curr Probl Cardiol, 2024; 49; 102858
20. Dhore-patil A, Modi V, Gabr E-M, Cardiac magnetic resonance findings in cardiac amyloidosis: Curr Opin Cardiol, 2024; 39; 395-406
21. Brownrigg J, Lorenzini M, Lumley M, Elliott P, Diagnostic performance of imaging investigations in detecting and differentiating cardiac amyloidosis: A systematic review and meta-analysis: ESC Heart Fail, 2019; 6; 1041-51
22. Scheel PJ, Mukherjee M, Hays AG, Vaishnav J, Multimodality imaging in the evaluation and prognostication of cardiac amyloidosis: Front Cardiovasc Med, 2022; 9; 787618
23. Porcari A, Masi A, Martinez-Naharro A, Redefining cardiac involvement and targets of treatment in systemic immunoglobulin AL amyloidosis: JAMA Cardiol, 2024; 9; 982
24. Adams D, Gonzalez-Duarte A, O’Riordan WD, Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis: N Engl J Med, 2018; 379; 11-21
25. Spoladore R, Falasconi G, Marcatti M, Advances in pharmacotherapy for cardiac amyloidosis: Expert Opin Pharmacother, 2021; 22; 469-81
26. Haslett JJ, Patel JK, Kittleson MM, Beta 2-microglobulin: Case report of a rare cause of cardiac amyloidosis: Eur Heart J Case Rep, 2023; 7; ytad239
27. Schuetz T, Schiller D, Klingel K, Unicentric Castleman’s disease associated with malignant cardiac Amyloid-A amyloidosis: A case report: Eur Heart J Case Rep, 2023; 7; ytad451
28. Nuzzi V, Porcari A, Gigli M, A case report of isolated cardiac light chain amyloidosis without clinically overt heart failure: An under-recognized presentation: Eur Heart J Case Rep, 2023; 7; ytad072
29. Milani P, Valentini V, Ferraro G, A patient with AL amyloidosis with negative free light chain results: Clin Chem Lab Med, 2016; 54; 1035-37
Figures
Figure 1. Echocardiographic findings showing restrictive filling pattern with E/E’: 19.88 and an E/A ratio >2 (arrow), a hallmark of cardiac amyloidosis.Figure 2. Echocardiographic finding showing right ventricle hypertrophy (arrow) and spontaneous echo contrast in left atrium.Figure 3. Echocardiographic finding showing left ventricular hypertrophy (arrow) with speckled myocardial appearance and pleural effusion.Figure 4. Echocardiographic finding showing systolic flow reversal (arrow) in the hepatic vein indicative of severe tricuspid regurgitation.Figure 5. Mild pericardial effusion and significant pleural effusion (arrow).Figure 6. Very low septal E’ (arrow).Figure 7. Diagnostic algorithmic approach for cardiac amyloidosis.Figure 8. Algorithms that summarize the approach used in treatment of cardiac amyloidosis. Transthyretin amyloidosis (ATTR); Heart failure (HF); Direct oral anticoagulant (DOAC); Cardiac variant transthyretin amyloidosis ATTRwt-CM; Variant transthyretin amyloidosis (ATTRv); Permanent pacemaker (PPM); Implantable cardioverter-defibrillator (ICD); Cardiac resynchronization therapy (CRT); Ventricular tachycardia (VT); Sudden cardiac death (SCD). In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952428
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952854
Most Viewed Current Articles
07 Dec 2021 : Case report  22,760,204
22,760,204
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,117
176,117
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,604
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,593
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133