11 March 2021: Articles 
A Woman with a 27-Year History of Hyperparathyroidism and Hypercalcemia Who Was Diagnosed with Low-Grade Parathyroid Carcinoma
Challenging differential diagnosis, Diagnostic / therapeutic accidents, Unusual setting of medical care, Rare disease, Educational Purpose (only if useful for a systematic review or synthesis)
Monika Kaszczewska1ABCDEF, Michał Popow2ABCDF, Witold Chudziński1ABCDEF*, Joanna Kaszczewska1BEF, Magdalena Bogdańska3BD, Joanna Podgórska4BC, Agnieszka Czarniecka5BC, Zbigniew Gałązka1ABCDEDOI: 10.12659/AJCR.930301
Am J Case Rep 2021; 22:e930301






Abstract
BACKGROUND: Parathyroid carcinoma (PC), accounting for 0.005% of all cancers, is responsible for less than 1% of all cases of primary hyperparathyroidism, and equally affects males and females, usually in 4th or 5th decades of life. PC can occur sporadically and can be associated with congenital genetic syndromes such as hyperparathyroidism-jaw tumor syndrome (HPT-JT), isolated familial hyperparathyroidism, or multiple endocrine neoplasia 1 and 2 syndromes. Surgery is the main treatment, with a limited role of radio- and chemotherapy, which allows 49-77% of patients to survive 10 years. In this work we report the case of a patient with parathyroid carcinoma, whose treatment required 13 surgeries over a period of 27 years, together with radiotherapy and pharmacological treatment.
CASE REPORT: A 51-year-old woman was first diagnosed with primary hyperparathyroidism in 1993 at the age of 23. From 1993 to present, she underwent 13 surgeries and 33 courses of radiotherapy due to recurrent lesions, which initially had a character of parathyroid adenomas, then parathyromatosis, and finally were diagnosed as parathyroid carcinoma. The patient also required and currently requires complex pharmacological treatment to control the calcemia and manage the complications of the primary disease. Supervision by the multidisciplinary professional medical team allows the patient to lead a normal life with good control of the disease.
CONCLUSIONS: Parathyroid carcinoma is a rare disease with a number of complications; however, obtaining satisfactory long-term survival with acceptable quality of life is achievable.
Keywords: Hyperparathyroidism, Primary, Parathyroid Neoplasms, Parathyroidectomy, Radiotherapy, Adjuvant, Hypercalcemia, Jaw Neoplasms, Quality of Life
Background
Parathyroid carcinoma (PC) is one of the rarest malignancies, accounting for only 0.005% of all cancers in North America and Western European countries, and accounting for less than 1% of all cases of primary hyperthyroidism (PHPT) [1–5].
PC typically appears during the mid-40s or 50s, with equal frequency in males and females. For comparison, parathyroid adenoma (PA) is more frequent in females (with sex ratio of 3: 1) and is diagnosed approximately 1 decade later compared to PC [1–3].
The pathogenesis of PC is unknown. It can occur as a sporadic disease or as part of genetic syndromes. Up to 15–24% of patients with hyperparathyroidism-jaw tumor syndrome (HPT-JT) develop PC. Among other genetic syndromes associated with PC, the following have been reported: multiple endocrine neoplasia types 1, 2, and 4 (MEN1, MEN2, MEN4) and isolated familial hyperparathyroidism (FIHP) [1–6]. Non-genetic risk factors include prior neck radiation, secondary and tertiary hyperparathyroidism (associated with renal failure), and thyroid cancer [1,3].
PC often has an indolent and progressive course. It is mainly hormonally functional with hypersecretion of parathyroid hormone (PTH), which causes symptoms related to severe hypercalcemia (eg, bone disease, renal failure, cardiac arrhythmia, and neurocognitive disturbances). Extremely rare cases of non-functional PC with normal serum calcium levels (accounting for up to 2% of all PCs) are also reported. Due to clinical symptoms similar to benign causes of hyperthyroidism, it is difficult to make a final diagnosis of PC, which is usually made postoperatively based on histological examination [3,6].
With very limited effectiveness of chemo- or radiotherapy, surgical treatment remains key in management of PC, with a reported 85% overall survival of patients at 5-year follow-up and 49–77% survival at 10-year follow-up [3,4,7,8].
The aim of this study was to present the case of 51-year-old woman in whom parathyroid adenoma and parathyromatosis preceded development of parathyroid cancer. During 27 years of treatment, she underwent 13 parathyroidectomies and 33 radiotherapy cycles of the neck and upper mediastinum.
Case Report
In 1992, a 23-year-old woman with previous history of several extracorporeal shock-wave lithotripsy (ESWL) procedures due to symptomatic nephrolithiasis, after a childbirth, presented with intense pain of the femur and muscle weakening. The patient was referred with a suspicion of myopathy to the Neurological Department, where hypercalcemia secondary to primary hyperparathyroidism (adenoma of the right lower parathyroid gland) was diagnosed. That year, the patient had a pathological fracture of right clavicle. In 1993, a right lower parathyroidectomy was performed and a partial resection of thyroid gland due to nodular lesions was conducted at the same time. In the 12 years of follow-up, the patient remained asymptomatic. However, in 2006 she was referred to the Department of Internal Diseases and Endocrinology due to symptoms of hyperparathyroidism manifesting with right upper-extremity pain. A recurrent lesion in the area of the previously removed right lower parathyroid gland was detected. Intraoperatively, a parathyromatosis was recognized. In subsequent years, there was a recurrence of the hyperparathyroidism, and growth of the adenomatous lesions significantly accelerated. Non-specifically, increased Ki-67 proliferation index (6%) in histopathological examination of parathyroid adenomas was reported. In 2010, 2013, and 2014, adenomatous parathyroid lesions were diagnosed in the right side of the neck with accompanying right-sided parathyromatosis (2014). A Technetium sestamibi scan (99m Tc-MIBI) showed a pathological collection of radionuclides below the jugular notch of the sternum. In the upper mediastinum, several lesions measuring from 21×29×34 mm to 7×5×7 mm with increased methoxyisobutylisonitrile (MIBI) collection (ectopic parathyroid tissue) were observed. At that time, the mutation of RET protooncogene (MEN 2) was excluded. The genetic and clinical analysis also excluded MEN1 syndrome, but further genetic tests were planned. Staining against parafibromin was negative (Figure 1). The ultrasound examination revealed advanced-stage nephrocalcinosis in the right kidney and lower pole of the left kidney. Due to the bell-shaped thorax detected in imaging examination, the suspicion of a bone structure development disorder was raised. Because of a hypercalcemic crisis (with calcium level approximately 4 mmol/l and parathormone [PTH] 7–23 times higher than normal values), the patient underwent resection of pathological parathyroid tissue as well as parathyromatosis located in muscles and adipose tissue. Initially, to control the calcium level, the patient received calcimimetics: cinacalcet with zoledronic acid and calcitonin (which was poorly tolerated and produced a skin rash). Due to limited efficacy of the above-mentioned treatment, the patient was switched to denosumab (a receptor activator of nuclear factor κ B-ligand [RANKL] inhibitor, a human monoclonal antibody usually used in the treatment of osteoporosis), which in this indication was used off-label, but this use is described in the medical literature.
In November 2015, the patient was diagnosed with another recurrent lesion, located between the right jugular vein and right common carotid artery, and again qualified for surgical treatment. After surgery, a significant (more than 50%) reduction in PTH concentration was observed. However, within 4 days, there was recurrence of hypercalcemia and a notable increase in PTH. Postoperatively, the parathyroid cancer was recognized in the histopathological examination, with 9 mitotic figures noted per 10 high-power fields, with negative immunochemical staining for chromogranin A and synaptophysin and Ki-67 of 7% to 10%. To control the level of calcium, the patient received 2 doses of denosumab during a period of 6 months, and received 500 units of vitamin D per day. Half a year later, in January 2016, the patient was admitted to the Internal Diseases and Endocrinology Department due to severe hypercalcemia and the threat of a hypercalcemic crisis. After a pharmacological treatment and stabilization of her condition, following oncological consultation, radiotherapy of the neck region was planned. At that time, germinal mutation of gene cell division cycle 73 (CDC73) was excluded.
In the following years, the recurrences of the lesions appeared more frequently, with shorter intervals between episodes, and was always located in the right cervical region (Figure 2).
In May 2016, the patient was again admitted to the hospital due to another hypercalcemic crisis, which was resistant to pharmacological management and required emergency surgical treatment. In the postoperative period, a pulmonary embolism developed in the artery supplying the 6th segment of the right lung. Despite resection of a 26-mm diameter tumor infiltrating the sternum manubrium, no normalization of PTH and calcium serum concentration was obtained; therefore, high doses of zoledronic acid and cinacalcet were added to the treatment. In the postoperative period, another 4 lesions were detected (3 in the cervical area and 1 located retroclavicularly), which were successfully removed in September 2016. A histopathological examination revealed a somatic mutation CDC73/HRPT2.
In 2017, regrowth of about 1-cm parathyroid tumors was observed at 2-month intervals, resulting in a hypercalcemic crisis. Due to lack of a satisfactory effect of surgical treatment (12 surgeries up to that time), radiotherapy (33 courses of conventional 2D radiotherapy of the neck and mediastinum region, with X-rays and a fractional dose of 2 Gy, for a total radiation dose of 66 Gy) was introduced following another surgical re-section of the lesions. The treatment was complicated with Staphylococcal sepsis originating from the catheter to the superior vena cava, which resulted in the patient being in a severe condition (3/4 points in Eastern Cooperative Oncology Group [ECOG] score), under hospice care, with a poor prognosis. After a few months of additional pharmacological treatment (cinacalcet and denosumab), the patient’s condition gradually improved until obtaining complete recovery. As before, a PTH-related hypercalcemia was observed, but it was well controlled (calcium levels above 3 mmol/l).
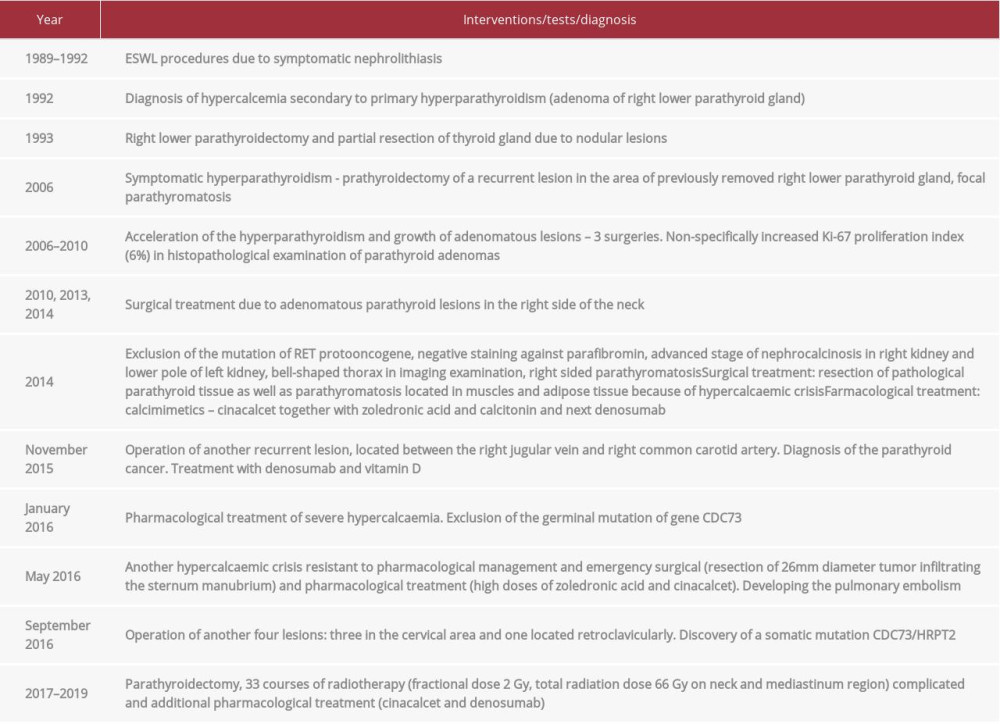
Following the treatment and recovery in subsequent hospitalizations, a PTH-related hypercalcemia was observed, without any metastatic lesions in imaging examinations. At present, the patient remains under direct medical supervision, with stable levels of PTH and mild hypercalcemia, well-controlled pharmacologically with cinacalcet and denosumab. As a result of a 27-year course of disease, the patient has a visible bell-shaped deformation of the thorax caused by multiple rib fractures secondary to parathyromatosis and accompanying calcium metabolism impairment, which is shown in Figure 3. Table 1 presents the highlights of diagnosis and treatment.
Discussion
Being one of the rarest cancers, parathyroid carcinoma accounts for only 0.005% of all malignancies, causing less than 1% of primary hyperparathyroidism [3,9,10]. To date, only about 1000 cases have been described in the medical literature [3,11]. Parathyroid cancer usually is discovered in the 5th and 6th decades of life (in patients age 45–59 years old), which is approximately 10 years before diagnosis of benign parathyroid tumors. The prevalence of parathyroid carcinoma is equal in males and females, unlike in parathyroid adenomas, which occur 3 times more often in females [3,12]. No ethnic or geographical differences were described regarding PC [3].
In our patient, parathyroid adenoma was diagnosed at the age of 23 and parathyroid cancer was confirmed in histopathology at the age of 46 (in 2015); however, the negative staining against parafibromin in 2014 might have suggested malignancy.
Parathyroid cancer is a disease of unknown pathogenesis. Most cases are sporadic forms, but familial occurrences were also described. In familial forms, PC may be associated with genetic syndromes, including hyperparathyroidism-jaw tumor syndrome (HPT-JT), isolated familial hyperparathyroidism, and MEN1 and MEN2 syndromes [3,6,13]. HPT-JT is a rare disease, inherited in an autosomal dominant pattern, featuring multiple parathyroid tumors and 15–24% risk of transformation into PC. It is connected with ossifying fibromas of the maxilla or mandible, renal abnormalities (in up to 50% of patients), and uterine neoplasms (in up to 75% of female patients) [3,6,13].
The most commonly observed mutations in PC are the ones of the tumor-suppressor CDC73 gene (at 1q31 loci) encoding parafibromin. These mutations, associated with lack or reduction of expression of parafibromin (a tumor-suppressor protein, controlling cell proliferation, apoptosis and DNA solidity), are associated with HPT-JT syndrome and are observed in up to 70% of sporadic forms of PC. Every third patient with PC has germline CDC73 gene mutation, suggesting that some patients with the sporadic form have the HPT-JT syndrome or its variant. In sporadic adenomas, CDC73 mutations are rarely observed, which suggests its limited role in the pathogenesis of PC [3,6,13]. On the contrary, modification in parafibromin expression appears in most PCs and infrequently in parathyroid adenomas, suggesting that detection of the CDC73 gene mutation combined with parafibromin expression loss might predict the probability of malignancy and poor clinical results [6,13]. Beside genetic factors, some authors indicated an association between PC and external radiation exposure or end-stage renal failure [6,14].
Typical pathological features of PC include large tumor size (diameter of 30 mm), capsular and blood vessel invasion, thick fibrous bands, trabecular growth pattern, cytologic atypia, tumor necrosis, and high mitotic activity (usually above 5 mitoses per 50 high-power fields). Immunohistochemistry (IHC) usually reveals positive immunostaining for PTH and chromogranin A. Malignancy in a parathyroid tumor is suggested by an elevated Ki-67 proliferation index (above 5%) [6,13].
In our case, a mutation of RET, MEN1 syndrome, and germinal mutation of gene CDC73 were excluded. Only a histopathological examination revealed a somatic mutation CDC73/HRPT2 in tumor tissue, consistent with negative staining against parafibromin. Postoperatively, the histopathological examination showed mitotic activity with 9 mitosis per 10 high-power fields and increased Ki-67 proliferation index (7% to 10%) with negative immunochemical staining for chromogranin A and synaptophysin. In our own experience, no strong correlation between MEN1 syndrome and PC was observed; in a group of over 40 individuals treated due to MEN1, none had parathyroid carcinoma. We would like to add that none of the patients from the group of more than 800 individuals with secondary or tertiary hyperparathyroidism were diagnosed with PC.
Due to the absence of clear diagnostic criteria, the confirmation of parathyroid cancer is difficult. Most of the diagnoses are made postoperatively based on histopathological examination; however, it is worth noting that currently there is no consensus concerning the pathological diagnostic criteria of PC [3,6,15,16]. Presence of metastases (nodal or distant) and invasion (vascular and adjacent tissue) are major criteria of malignancy that appear at the end stage of the disease in less than 10% of patients [6,13]. Clinical features and symptoms suggesting PC include distinctly increased plasma levels of PTH (3–10 times above normal values) and calcium (usually above 14 md/dl or 3.5 mmol/l), large size of parathyroid lesion (>3 cm), male sex, relatively young age, severe kidney and skeletal manifestations, neurological signs, palpable cervical mass, and laryngeal nerve palsy. The so-called >3+>3 rule (more than 3-cm diameter together with more than 3 mmol/l serum calcium concentration) suggested by Talat and Schulte is an aid in PC diagnosis [3,6,13,17].
Due to its indolent course, most PCs have similar, but more severe, clinical manifestations compared to benign parathyroid tumors. Up to 80–90% of patients experience renal impairment (eg, renal failure, nephrolithiasis, nephrocalcinosis, osteitis fibrosa cystica) or bone involvement (eg, osteopenia, pathologic fractures, bone pain). Other symptoms associated with hypercalcemia are fatigue, malaise, polydipsia, polyuria, weight loss, confusion, and gastrointestinal manifestations (eg, anorexia, nausea, vomiting, abdominal pain, peptic ulcer, pancreatitis) [6,13]. Some authors reported very rare cases of nonfunctioning PC with symptoms limited to those of local growth and invasion (eg, neck mass, hoarseness, dysphagia) [6,8,18].
Our patient had repeated hypercalcemic crises with calcium level above 3 mmol/l and elevated PTH level (around 7 to 23 times above reference range values). Reported hypercalcemic symptoms include nephrolithiasis, bones pain, and muscle weakness. The diameters of the tumors were up to 3.5 cm.
Patients with PC are at risk of hypercalcemic crisis with serum calcium level above 16 mg/dl (3.99 mmol/l). This condition is an acute medical emergency. It occurs more often in functioning PCs than in benign parathyroid tumors. Symptoms include impaired consciousness, nausea, vomiting, abdominal pain, arrhythmia, polyuria, and dehydration accompanying severe hypercalcemia. Medical treatment decreasing serum calcium and improving metabolic disturbances is key in stabilization of patients awaiting surgery as well as in inoperable cases. The main pharmacological management is hydration with intravenous saline infusion, which helps to restore intravascular volume and renal diuresis. After acquiring euvolemia, to inhibit calcium re-absorption, loop diuretics are often used. In case of failure of pharmacological calcium-lowering treatment, hemodialysis can be introduced. In most cases, further pharmacological treatment with calcitonin, calcimimetic agents (cinacalcet), and agents that block bone resorption (bisphosphonates, denosumab) is required.
Bisphosphonates (eg, pamidronate, zoledronic acid) inhibit osteoclastic activity, but the effects of treatment are visible after longer periods of time (3–5 days). The limitation in the use of bisphosphonates is glomerular filtration rate (GFR) >30 ml/min/1.73 m2. Denosumab, a human monoclonal antibody against the RANK ligand, limiting bone resorption by osteoclasts, is used for patients resistant to bisphosphonates in the treatment of malignancy with persistent hypercalcemia. Both of these agents can produce mandibular osteonecrosis [19,20]. In cases requiring rapid calcium decrease, calcitonin may be used to prevent bone resorption and decrease renal tubular calcium reabsorption. It occasionally can cause anaphylactic shock in patients with allergy to salmon. Cinacalcet, an allosteric modulator of the calcium-sensing receptor (CaSR) on the membrane of parathyroid cells, increasing receptor’s affinity for calcium and decreasing secretion of PTH, is the most effective treatment in inoperable PC and can normalize calcium levels in up to 66% of patients [3,6,13]. However, it should be noted that decreased CaSR expression was observed in some patients with severe course of parathyroid carcinoma [21].
In our case, in over 27 years of follow-up, we observed recurrences of the hypercalcemic crisis, which appeared more frequently in later phases of the disease and at shorter intervals. Persistent and symptomatic hypercalcemia was one of the greatest therapeutic challenges. To control calcium levels, the patient was treated with forced diuresis (saline infusions and after obtaining euvolemia loop diuretics) and cinacalcet, together with zoledronic acid. Calcitonin was applied occasionally due to in tolerance and occurrence of skin rash. Because of the limited efficacy of bisphosphonate and cinacalcet, the patient received denosumab, which resulted in normalization of calcium level. There was no mandibular osteonecrosis as an adverse effect of simultaneous treatment with zolendronic acid and denosumab.
For detecting parathyroid anomalies in patients with primary hyperparathyroidism, ultrasonography (US) and 99mTc-sestamibi scintigraphy (MIBI) imaging are routinely used. Those radiographic methods are used for localizing enlarged parathyroid glands [3,13]. Recently, some research found higher retention levels of MIBI in PCs than in benign parathyroid tumors [22]. For localization the parathyroid mass, invasion of surrounding tissues, and distant metastases, 4-dimensional computed tomography (4D CT) and magnetic resonance imaging (MRI) scans are used [3,13]. Other methods for preoperative localization of parathyroid tissues include positron emission tomography/computed tomography (PET/CT) with 11C-methionine or 18-fluorocholine [23,24]. Despite relatively high sensitivity and specificity, those methods are not routinely used in diagnosis due to limited availability and very high costs. Another method which might be used in the diagnosis in neuroendocrine tumors (NETs) is (68)Ga-DOTATATE PET/CT. For parathyroid lesions, this method has sensitivity and specificity of 28% and 100%, respectively. This method was not used in our patient [25]. When PC is suspected, fine-needle aspiration biopsy (FNAB) is not recommended due to its limited diagnostic value in this indication and the risk of rupture of the lesion capsule and potential spread of tumor cells. FNAB can be useful for the confirmation of metastatic tissue and PTH-secretion (PTH aspirate-hormone test) [3,13].
In our patient, ultrasound, CT scans, and MIBI were used for confirmation of the localization of parathyroid cancer and metastases. The recommended treatment (first-line therapy) for PC is complete surgical resection with microscopically negative margins, as well as en-bloc resection of the primary lesion with ipsilateral thyroid lobe and adjacent involved structures (recurrent laryngeal nerve, the trachea, and/or the esophageal wall) with avoidance of spreading neoplastic cells. Preoperative diagnosis and intraoperative recognition, as well as surgeon experience, results in an adequate surgical approach [3,6,13].
In up to 49–60% of patients, another surgical resection is usually needed after 2–3 years due to recurrence of parathyroid cancer lesions and severe hypercalcemia. Because reoperation is infrequently curative and recurrences are common, patients require frequent, life-long check-ups to detect it at an early stage [3,6,13,26].
PC is generally regarded as radiotherapy-resistant, so the role of postoperative adjuvant radiation therapy (RT) is controversial and not widely used. Some studies have suggested a potential benefit of postoperative RT (doses 40–70 Gy) in high-risk cases, resulting in reduction of cancer recurrence [3,6,8,13,27].
In our patient, because of the insufficient effect of pharmacological and surgical treatment (13 surgeries), 33 courses of palliative radiotherapy were administered, which resulted in stabilization of clinical course of the disease in terms of prolonging intervals between recurrences. No another surgical procedure was required following the radiotherapy.
Due to low efficacy, chemotherapy is rarely applied in the treatment of PC and no standard chemotherapy protocols are available. Several studies reported use of dacarbazine, fluorouracil, cyclophosphamide, methotrexate, doxorubicin, and lomustine. Sorafenib is occasionally used as targeted therapy against PC. Possible use of chemotherapy as adjuvant treatment in PC must be individually considered [3,6,13]. Chemotherapy was not applied in our patient’s treatment.
Other experimental treatments, which require further studies, include radiofrequency ablation and arterial embolization of the tumor or metastases, as well as immunotherapy with human and bovine PTH peptides and dendritic cells [3,6,15].
Because of the indolent process of PC, overall 5- and 10-year survival rates are reported as 78–91% and 49–77%, respectively. Persistent neoplasm or recurrences are observed in 23–51% of cases; they are manifested by symptoms related to hypercalcemia, and they are also caused by loco-regional recurrence and distant metastases, usually in the lungs, bones, and liver. Recurrences appear after 2.5 to 5 years following first surgery; however, a latency period of 23 years was also reported. Mortality is related to uncontrollable hypercalcemia with subsequent organ impairment, such as renal failure. Due to difficulty in detecting all lesions, up to 40% of patients undergo incomplete lesion resection. There is no consensus concerning prognosis; however, some studies indicate a negative impact of factors such as: age, sex, metastasis, time to recurrence, high serum calcium concentration at recurrence, number of recurrences, number of calcium-lowering drugs, or inability to achieve complete tumor resection. These observations come mainly from single-center studies based on relatively small samples [3,13].
Current evidence on the connection between parathyroid adenoma and parathyroid carcinoma is questionable. Retrospective population studies in Finland (2000–2010) and Sweden (1958–2008) reported a correlation between parathyroid adenoma and parathyroid carcinoma; however, no evidence of malignant transformation of adenoma was found [6,28,29].
This observation is consistent with the disease progression in our patient, in whom the diagnosis of parathyroid carcinoma was made approximately 23 years after initial primary hyperparathyroidism due to parathyroid adenoma, and 2 years after the diagnosis of parathyromatosis. Because of lack of unified diagnostic criteria for PC, confirming the pathology remains challenging. Immunostaining for parafibromin, which revealed normal expression of this protein in normal cells and absence in carcinomatous cells, together with genetic studies, suggests monoclonal proliferation of parathyroid carcinoma, tumor spread, and, finally, metabolic consequences related to the mass effect.
In our opinion, there might be a connection between PC and parathyromatosis, but no such cases were described in the literature and eventual confirmation of this theory needs further studies.
Conclusions
The clinical presentation and the character of symptoms might be the driver for intensifying diagnostic processes in patients with hyperparathyroidism. In case of the recurrence of parathormone-related hypercalcemia, together with recurrent lesion in the same localization, one should consider parathyroid carcinoma, regardless of previous histopathological examination results indicating a benign process.
Parathyroid carcinoma is one of the rarest malignancies, associated with a number of complications mainly related to hypercalcemia, in which obtaining satisfactory long-term survival with acceptable quality of life is achievable.
Figures
References:
1.. Salcuni AS, Cetani F, Guarnieri V, Parathyroid carcinoma: Best Pract Res Clin Endocrinol Metab, 2018; 32(6); 877-89
2.. Campennì A, Giovinazzo S, Pignata SA, Association of parathyroid carcinoma and thyroid disorders: A clinical review: Endocrine, 2017; 56(1); 19-26
3.. Wei CH, Harari A, Parathyroid carcinoma: Update and guidelines for management: Curr Treat Options Oncol, 2012; 13(1); 11-23
4.. Lee PK, Jarosek SL, Virnig BA, Trends in the incidence and treatment of parathyroid cancer in the United States: Cancer, 2007; 109(9); 1736-41
5.. Sadler C, Gow KW, Beierle EA, Parathyroid carcinoma in more than 1,000 patients: A population-level analysis: Surgery, 2014; 156(6); 1622-29 ; discussion 1629–30
6.. Cetani F, Pardi E, Marcocci C, Parathyroid carcinoma: Front Horm Res, 2019; 51; 63-76
7.. Busaidy NL, Jimenez C, Habra MA, Parathyroid carcinoma: A 22-year experience: Head Neck, 2004; 26(8); 716-26
8.. Harari A, Waring A, Fernandez-Ranvier G, Parathyroid carcinoma: A 43-year outcome and survival analysis: J Clin Endocrinol Metab, 2011; 96(12); 3679-86
9.. Dudney WC, Bodenner D, Stack BC, Parathyroid carcinoma: Otolaryngol Clin North Am, 2010; 43(2); 441-53
10.. Sandelin K, Auer G, Bondeson L, Prognostic factors in parathyroid cancer: A review of 95 cases: World J Surg, 1992; 16(4); 724-31
11.. Givi B, Shah JP, Parathyroid carcinoma: Clin Oncol (R Coll Radiol), 2010; 22(6); 498-507
12.. Shane E, Clinical review 122: Parathyroid carcinoma: J Clin Endocrinol Metab, 2001; 86(2); 485-93
13.. Rodrigo JP, Hernandez-Prera JC, Randolph GW, Parathyroid cancer: An update: Cancer Treat Rev, 2020; 86; 102012
14.. Ságová I, Stančík M, Kentoš P, [Parathyroid cancer]: Vnitr Lek, 2017; 63(2); 139-44 [in Czech]
15.. Cetani F, Pardi E, Marcocci C, Parathyroid carcinoma: A clinical and genetic perspective: Minerva Endocrinol, 2018; 43(2); 144-55
16.. Shane E, Clinical review 122: Parathyroid carcinoma: J Clin Endocrinol Metab, 2001; 86(2); 485-93
17.. Schulte KM, Talat N, Diagnosis and management of parathyroid cancer: Nat Rev Endocrinol, 2012; 8(10); 612-22
18.. Cetani F, Frustaci G, Torregrossa L, A nonfunctioning parathyroid carcinoma misdiagnosed as a follicular thyroid nodule: World J Surg Oncol, 2015; 13; 270
19.. Filleul O, Crompot E, Saussez S, Bisphosphonate-induced osteonecrosis of the jaw: A review of 2,400 patient cases: J Cancer Res Clin Oncol, 2010; 136(8); 1117-24
20.. Qi WX, Tang LN, He AN, Risk of osteonecrosis of the jaw in cancer patients receiving denosumab: A meta-analysis of seven randomized controlled trials: Int J Clin Oncol, 2014; 19(2); 403-10
21.. Witteveen JE, Hamdy NA, Dekkers OM, Downregulation of CASR expression and global loss of parafibromin staining are strong negative determinants of prognosis in parathyroid carcinoma: Mod Pathol, 2011; 24(5); 688-97
22.. Zhang M, Sun L, Rui W, Semi-quantitative analysis of 99mTc-sestamibi retention level for preoperative differential diagnosis of parathyroid carcinoma: Quant Imaging Med Surg, 2019; 9(8); 1394-401
23.. Braeuning U, Pfannenberg C, Gallwitz B, 11C-methionine PET/CT after inconclusive 99mTc-MIBI-SPECT/CT for localisation of parathyroid adenomas in primary hyperparathyroidism: Nuklearmedizin, 2015; 54(1); 26-30
24.. Kim SJ, Lee SW, Jeong SY, Diagnostic performance of F-18 fluorocho-line PET/CT for parathyroid localization in hyperparathyroidism: A systematic review and meta-analysis: Horm Cancer, 2018; 9(6); 440-47
25.. Lastoria S, Marciello F, Faggiano A, Role of (68)Ga-DOTATATE PET/CT in patients with multiple endocrine neoplasia type 1 (MEN1): Endocrine, 2016; 52(3); 488-94
26.. Kebebew E, Arici C, Duh QY, Clark OH, Localization and reoperation results for persistent and recurrent parathyroid carcinoma: Arch Surg, 2001; 136(8); 878-85
27.. Christakis I, Silva AM, Williams MD, Postoperative local-regional radiation therapy in the treatment of parathyroid carcinoma: The MD Anderson experience of 35 years: Pract Radiat Oncol, 2017; 7(6); e463-70
28.. Ryhänen EM, Leijon H, Metso S, A nationwide study on parathyroid carcinoma: Acta Oncol, 2017; 56(7); 991-1003
29.. Fallah M, Kharazmi E, Sundquist J, Hemminki K, Nonendocrine cancers associated with benign and malignant parathyroid tumors: J Clin Endocrinol Metab, 2011; 96(7); E1108-14
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report  22,759,844
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133