13 May 2021: Articles 
Delayed Severe Hemolytic Transfusion Reaction During Pregnancy in a Woman with β-Thalassemia Intermediate: Successful Outcome After Eculizumab Administration
Unusual or unexpected effect of treatment, Diagnostic / therapeutic accidents, Rare coexistence of disease or pathology
Giovanna Cannas1ABCDEFG*, Léa Dubreuil2BD, Axel Fichez3BD, Mathieu Gerfaud-Valentin4BD, Anne-Lise Debard2BD, Arnaud Hot1DDOI: 10.12659/AJCR.931107
Am J Case Rep 2021; 22:e931107






Abstract
BACKGROUND: Delayed hemolytic transfusion reactions (DHTR) are life-threatening complications mostly triggered by red blood cell (RBC) transfusions in patients with hemoglobinopathy.
CASE REPORT: We present a case of DHTR and hyperhemolysis syndrome in a 39-year-old pregnant woman with a history of β-thalassemia intermediate in whom the hemoglobin (Hb) level fell to 27 g/L after transfusion of 2 units of crossmatch-compatible packed RBCs. No allo- or auto-antibody formation was detected. Administration of intravenous immunoglobulins and methylprednisolone followed by anti-CD20 rituximab was tried, but was unsuccessful. Infusions of eculizumab (900 mg twice at a 7-day interval) followed by another course of intravenous immunoglobulins (2 g/kg/day for 5 days) and combined with repeated erythropoietin injections (darbepoetin alpha 300 µg/week) finally allowed biological and clinical improvement. Blood counts remained controlled until delivery. Despite signs of intrauterine growth retardation, she gave birth by cesarean section at 31 weeks of pregnancy to a 1.15-kg infant.
CONCLUSIONS: Eculizumab seems to be of benefit in DHTR associated with hyperhemolysis and should be used early in the treatment of this pathology. Despite premature birth, our case report showed an acceptable outcome for the infant when eculizumab treatment was used during pregnancy.
Keywords: Beta-thalassemia, eculizumab, Pregnancy, Transfusion reaction, Antibodies, Monoclonal, Humanized, Cesarean Section, Isoantibodies
Background
Delayed hemolytic transfusion reactions (DHTR) are rare. They are potentially life-threatening complications mostly triggered by red blood cell (RBC) transfusions in patients with sickle cell disease (SCD) [1,2], but have been occasionally described in patients with other hematological conditions [3–5] such as thalassemia [6–10]. The main cause is alloimmunization, but the pathogenic mechanisms are complex and evidence of alloimmunization is not always found [11–13].
We report here data from a pregnant woman with β-thalassemia intermediate hospitalized for severe DHTR and hyper-hemolysis, following crossmatch-compatible RBC transfusion. She was successfully treated with eculizumab in addition to intravenous immunoglobulins and erythropoietin (EPO).
Case Report
A 39-year-old woman with a history of β-thalassemia intermediate (diagnosed at the age of 10 years as a frame shift mutation involving the codon 6 of β-globulin) with no evidence of combined glucose-6-phosphate dehydrogenase deficiency was seen on 1 April 2019 at 17 weeks of a first pregnancy. Treatment with hydroxyurea and deferasirox was stopped when pregnancy was planned (2018). Her hemoglobin (Hb) level was 76 g/L. Three weeks later, her Hb level dropped to 71 g/L and, because she had to travel abroad for 1 week, transfusion support [2 units of crossmatch-compatible packed RBCs after antibody screening confirmed the presence of 3 known antibodies: anti-FY1 (Fya), anti-JK1 (Jka), and anti-MNS3 (S)] was performed without any complications. Her past history revealed a total of 5 prior RBC transfusions, of which the last was administered 6 years ago. Her blood group was A Rh+, extended RBC phenotype C–, E+, c+, e–, K–. She presented again at our consultation on 11 May. At that time, she was deeply jaundiced with severe asthenia, dyspnea, and headaches, and noticed dark-colored urine. Her Hb level had dropped to 41 g/L. Vital signs remained otherwise normal. Laboratory findings were consistent with acute hemolysis with serum bilirubin at 30 µmol/L, unconjugated bilirubin at 20 µmol/L, lactate dehydrogenase (LDH) level at 943 UI/L (normal range: 125-220 UI/L), and undetectable haptoglobin. Direct antiglobulin test (DAT) was negative, as were cultures of blood and urine. Alloantibodies against JK1 titrated at 8. No previously undetected RBC alloantibodies could be identified on repeated serum samples. Although the patient was transfused with group O RBCs, no double cell population was found when determining blood grouping, attesting to a potential hemolysis of the transfused RBCs. The ultrasound showed a fetus with good vitality, no morphological abnormalities, but with cranial and abdominal biometrics to be monitored (suspicion of intrauterine growth retardation).
She was hospitalized and placed on steroids (1 mg/kg per day) and recombinant EPO injections (methoxy polyethylene glycol-epoetin beta 360 mg for 4 days) combined with folic acid (30 mg/day) and iron supplementation (ferric hydroxide injection 100 mg). She remained hemodynamically stable, nonpyretic, and eupneic (saturation at 97% on oxygen therapy). Because her Hb level dropped to 33 g/L, a crossmatch-compatible RBC transfusion of 2 units was attempted, leading to a Hb level of 47 g/L, which dropped again to 31 g/L with signs of hemolysis 24 h later. Because of a potential thrombotic micro-angiopathy (2% of schizocytes and low platelet count), plasma exchanges were then attempted but stopped after 2 sessions because of a low hematocrit level (<20%). Intravenous immunoglobulins (2 g/kg total dose on days 1 to 5) were then administered and methylprednisolone was given at 250 mg/day followed by the administration of anti-CD20 antibody rituximab (375 mg/m2 on days 1, 4, 8, and 12). Despite those treatments, Hb level remained low, with a nadir at 27 g/L. The pregnancy was regularly monitored by ultrasound (22 May) confirming fetal vitality but with signs of intrauterine growth retardation and fetal anemia without oligo- or hydramnios. Because of reticulocytopenia, treatment with repeated EPO injections was intensified (epoetin alpha 50 UI/Kg×3 per week then darbepoetin alpha 300 µg/week). Bone marrow (BM) aspirate (15 May) showed erythroblastic hyperplasia that was contradicting with the low reticulocyte count in peripheral blood. BM aspirate eliminated medullary necrosis and macrophage activation syndrome. Megakaryocytes were present without morphological abnormality, indicating a peripheral origin of the thrombopenia. The ultrasound confirmed the presence of large splenomegaly (25 cm), involved in the thrombocytopenia (nadir at 52 G/L). In the absence of peripheral blood improvement, on 5 June the patient received, after meningococcal and pneumococcal vaccinations, infusions of eculizumab (900 mg on days 1 and 7) and again administration of intravenous immunoglobulins (2 g/kg/day for 5 days) on 12 June, leading to biological improvement (Hb increased in concentration to 65 g/L, platelets to 162 G/L, reticulocytes to 76 G/L, and haptoglobin to 0.26 g/L) (Figure 1), allowing transfer of the patient to the Obstetrics Department on 3 July. Blood counts remained controlled until delivery, performed on 12 July by cesarean section (31 weeks of pregnancy). The newborn (male sex, weight at birth 1.15 kg, hemoglobin at 146 g/L, DAT-negative) was hospitalized in the Neonatal Intensive Care Unit because of severe intrauterine growth retardation. Hemorrhage at delivery was about 350 mL. The mother’s Hb levels before and after delivery remained stable (63 g/L). Post-delivery complications were simple, only consisting in early pyelonephritis at
Screening studies, performed in order to explain the hyperhemolysis, included searches for novel anti-erythrocytic antibodies, and explorations in blood donors. DAT, which was initially negative, was positive from 15 May for C3d only, and increased progressively. It finally decreased in intensity after 3 June, to reach negativity again on 13 August. The donors KIDD system genotyping found JK*2/*2, clearly reflecting the expected phenotype of JK: -1;2. To eliminate the presence of an anti-private antibody in the recipient, compatibility tests were performed between donor and recipient serum, but results were negative. Father genotype was also performed during pregnancy, showing a JK*2/*2 genotype. At birth, the infant was effectively Jka–. Absorption tests on homologous RBCs were also carried out from 12 May, and did not show any alloantibodies of current specificity. A direct elution test showed no evidence of antibodies on the patient RBC surface. It was hypothesized that all RBCs that have bound antibodies were already hemolyzed. The presence of anti-KEL6, anti-RH20, anti-RH23, or antiYT2 was also eliminated. The presence of an anti-LU1 was also ruled out, as many anti-private antibodies by using RBCs presenting low-frequency antigens (DI3 MNS9, SC2, KEL6, RH20, RH23, Bga, Bgb, and Bgc).
Discussion
Our case report highlights the potential severity of DHTR with hyperhemolysis in a patient with β-thalassemia intermediate. This situation occurring in the setting of pregnancy was particularly life-threatening both for the mother and the fetus. Management of pregnant women with blood disorders is always challenging [14–16]. Each patient should be examined individually considering both aggressiveness of the disease and the stage of the pregnancy when a therapy is applied. Here, a collaboration is needed among the patient and her family, hematologists, and obstetricians.
Patients with DHTR demonstrated persistent RBC hemolysis involving both donor RBCs and the patient’s own RBCs [17]. Subsequent transfusions undergo rapid destruction that is not prevented by the use of antigen-negative crossmatch-compatible RBCs. Various proposed mechanisms have been suggested. Prior studies support the idea of a dual hemolytic response against both transfused and autologous RBCs, suggesting that the immune-mediated hemolysis results from a defect in complement regulation [18,19]. Another mechanism could involve the destruction of RBCs by macrophages through interactions between macrophage adhesion molecules and RBC surface glycoproteins [20,21]. An inappropriately low reticulocyte count has often been reported, suggesting a transfusion-induced suppression of erythropoiesis [22]. Hyperhemolysis generally occurs 5 to 10 days after transfusion, but can also be seen until 3 weeks after transfusion [23]. In one-third of DHTR cases, no antibodies can be detected [12,13]. This could be explained by a lack of sensitivity of current immunohematological techniques, but also by a concomitant underlying inflammatory mechanism [23,24]. Positivity of DAT has been previously reported in relationship with the length of the interval between transfusion and hemolysis: it is often negative if less than 7 days and positive if more than 7 days [12,25]. When present, alloantibodies appear between days 4 and 7 after transfusion and reach their maximum titration between days 10 and 15. Because in some patients the antiglobulin reactions due to an anti-complement remained detectable on circulating RBCs for months, it was hypothesized that complement is regularly activated in DHTR and binds not only to donated, but probably also to autologous RBCs [26]. Altered membrane phospholipid distribution on RBCs seems to be sufficient to trigger complement activation, then opsonization, accelerated eryptosis of donor RBCs, and eryptosis of recipient RBCs, as a consequence of donor RBCs hemolysis [27,28]. A mechanism of phosphatidylserine exposure induced by platelet-activating factor secretion during DHTR was recently proposed to explain innocent bystander hemolysis [29].
In our case report, it could be hypothesized, from previous publications, that the reactivity of the previously formed anti-Jka alloantibody would have been resumed (via an unidentified mechanism) despite the transfusion of Jka– RBCs. Anti-Jka auto-antibodies, mimicking the specificity of the anti-alloanti-bodies, could have been formed to exert the same hemolytic activity as alloantibodies against the patient’s RBCs by activating the complement. This is supported by the clinical improvement concomitant with DAT negativization after eculizumab therapy.
Beside its potential mechanism of action, our case report raises several notable issues: (i) it confirms that DHTR with hyper-hemolysis is not restricted to patients with SCD; (ii) it demonstrates that eculizumab therapy combined with EPO is of potential efficacy in this setting; and (ii) it shows that pregnancy could be brought to term with fetal normal vitality despite the very low Hb levels.
To our knowledge only a few prior publications have reported DHTR followed by life-threatening hyperhemolysis in patients with thalassemia [6–10], and none occurring during pregnancy. Among the 4 cases available on Medline, one reports a 2-year-old boy with β-thalassemia major who developed, after several transfusions, hyperhemolysis with a positive (C3d only) DAT and no significant RBC allo- or auto-antibodies [6]. Another publication reports a 1.5-year-old girl with homozygous β-thalassemia who developed hyperhemolysis after a first transfusion [7]. Serologic analyses showed a positive DAT with anti-complement (C3d), but no results with anti-immunoglobulins. A third case involved a 1-year-old girl with β-thalassemia major who developed hyperhemolysis following an initial pheno-typically-matched blood transfusion [8]. The DAT was strongly positive with C3d and immunoglobulin-G. A weak anti-H and a weak cold auto-antibody were also detected. The last case reports a 73-year-old man with β-thalassemia intermediate who developed DHTR after a third RBC crossmatch-compatible transfusion [10]. Three alloantibodies (anti-N, anti-S, and anti-K) and a monospecific autoanti-JKb were identified.
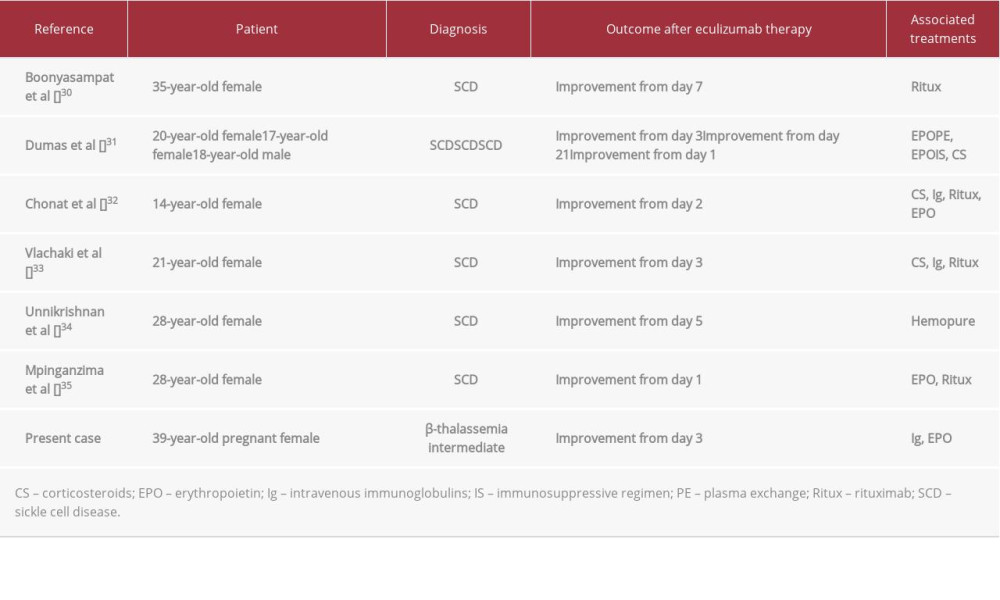
No optimal treatment has currently been established for DHTR. Transfusion should be avoided unless absolutely clinically necessary [19]. Some treatments have been reported to offset hemolysis, including steroids, intravenous immunoglobulins, or rituximab. However, in the present case, they did not adequately block hemolysis and did not help patient recovery. Steroids are known to be ineffective on cold agglutinin hemolysis and have no effect on RBC membrane complement reaction. Intravenous immunoglobulins are often ineffective [25]. Rituximab, proposed in cold agglutinin hemolysis and as a prophylactic agent, may prevent macrophage-dependent lysis of RBCs and reticulocytes [30]. It has been shown to prevent at least the occurrence of newly formed antibodies and to minimize the risk of severe DHTR [31]. However, recurrent hyperhemolytic syndrome is extremely rare and there is no way to predict which patients will have a recurrence after blood transfusion [32]. Furthermore, although rituximab treatment has been shown to be effective in DHTR [33,34], it must be pointed out that patients should be closely followed up due to the prolonged immunosuppression after rituximab, which can expose them to risk of severe infections. Complement activation may be involved in DHTR [35]. The deleterious effect of free heme on endothelial cells may also trigger the alternative complement activation [36]. Recently, eculizumab, an anti-C5 monoclonal antibody targeting complement activation and therefore inhibiting formation of the membrane attack complex, has been proposed to treat DHTR with hyperhemolysis in patients with SCD [37–42], ABO-incompatible RBC transfusion [43], or severe immunocompromised status [5]. Eculizumab demonstrated a high efficacy in patients with hemoglobinopathy (Table 1). Despite its efficacy, infusion reactions have been reported and eculizumab has been shown to increase the risk of infection by encapsulated organisms, leading ideally to a meningococcal vaccination at least 2 weeks prior the initiation of therapy [44].
Pregnancy in women with hemoglobinopathy may be associated with severe adverse fetal outcome and therefore poses a difficult challenge for both the patient and the attending physician. In case of planned pregnancy, risk/benefit evaluations must be carried out on an individual basis with careful counseling of both parents. Although uncommon, DHTR with hyperhemolysis represents a major complication in these patients and, in case of pregnancy, could be life-threatening for both mother and fetus in the absence of rapid efficient therapy. Data on the use of eculizumab in women during pregnancy are scarce. Despite premature birth, our case report showed an acceptable outcome for the infant when eculizumab treatment was used during pregnancy. Because complement dys-regulation has been reported in pregnant women during the third trimester and in women with preeclampsia, eculizumab may be a useful agent in other high-risk situations such as hyperhemolysis [45]. A series of 75 pregnancies in 61 women showed the benefit of eculizumab for women with paroxysmal nocturnal hemoglobinuria, as evidenced by a high rate of fetal survival and a low rate of maternal complications [46]. The study suggested that eculizumab can cross the placenta at low levels, but the drug was not detected in the breast milk samples. Interestingly, the novel long-acting C5 inhibitor ravulizumab was shown to be non-inferior to eculizumab for all studied endpoints, including intravascular hemolysis in paroxysmal nocturnal hemoglobinuria [47]. Furthermore, a recent evaluation showed a total incremental cost substantially lower for the novel agent versus eculizumab [48].
Conclusions
While additional immunosuppressive therapies and EPO could have affected the patient outcome, the rapid response after eculizumab therapeutic introduction suggests its ability to block the consequences of alternative complement pathway. Known for its rapid resolution of hemolysis [49], eculizumab seems therefore of potential benefit in DHTR associated with hyper-hemolysis. It should therefore be used earlier in the treatment of this pathology, especially in severe forms, tailored by a multidisciplinary approach involving hematologists, hemo-pathologists, and blood bank centers. Clinical trials are warranted and biological studies are needed to better explore the mechanisms of action of DHTR and the potential benefit of prophylactic complement inhibition in patients at risk.
References:
1.. Ness PM, Shirey RS, Thoman SK, Buck SA, The differentiation of delayed serologic and delayed hemolytic transfusion reactions: Incidence, long-term serologic findings, and clinical significance: Transfusion, 1990; 30; 688-93
2.. Vidler JB, Gaedner K, Amenyah K, Delayed haemolytic transfusion reaction in adults with sickle cell disease: A 5-year experience: Br J Haematol, 2015; 169; 746-53
3.. Treleaven JG, Win N, Hyperhaemolysis syndrome in a patient with myelofibrosis: Hematology, 2004; 9; 147-49
4.. Rogers M, Smith G, Hyperhaemolysis in a patient with chronic lymphocytic leukaemia: Transfus Med, 2014; 24; 123-24
5.. Gupta S, Fenves A, Nance ST, Sykes DB, Dzik WS, Hyperhemolysis syndrome in a patient without a hemoglobinopathy, unresponsive to treatment with eculizumab: Transfusion, 2015; 55; 623-28
6.. Morawakage LR, Perera BJ, Dias PD, Hyperhemolysis in a patient with beta-thalassemia major: Asian J Transfus, 2009; 3; 26-27
7.. Hannema SE, Brand A, van Meurs A, Delayed hemolytic transfusion reaction with hyperhemolysis after first red blood cell transfusion in child with beta-thalassemia: Challenges in treatment: Transfusion, 2010; 50; 429-32
8.. Grainger JD, Marker Y, McManus A, Wynn R, Refractory hyperhaemolysis in a patient with β-thalassemia major: Transfus Med, 2001; 11; 55-57
9.. Kashyap R, Choudhry VP, Delayed hemolytic transfusion reaction in a thalassemic child: Indian J Pediatr, 1994; 61; 726-28
10.. Dolatkhah R, Esfahani A, Torabi SE, Delayed hemolytic transfusion reaction with multiple alloantibody (anti S, N, K) and a monospecific auto-anti-JKb in intermediate β-thalassemia patient in Tabriz: Asian J Transfus Sci, 2013; 7; 149-50
11.. Yazdanbakhsh K, Ware RE, Noizat-Pirenne F, Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management: Blood, 2012; 120(3); 528-37
12.. Talano JAM, Hillery CA, Gottschall JL, Delayed hemolytic transfusion reaction/hyperhemolysis syndrome in children with sickle cell disease: Pediatrics, 2003; 111; e661-65
13.. De Montalembert M, Dumont MD, Heilbronner C, Delayed hemolytic transfusion reaction in children with sickle cell disease: Haematologica, 2011; 96; 801-7
14.. Karagiannis P, Alsdorf W, Tallarek AC, Treatment of refractory acute myeloid leukaemia during pregnancy with venetoclax, high-dose cytarabine and mitoxantrone: Br J Haematol, 2021; 192; e60-63
15.. Galiba Atipo Tsiba FO, Itoua C, Pregnancy outcomes among patients with sickle cell disease in Brazzaville: Anemia, 2020; 2020; 1989134
16.. Sahu KK, Dhibar DP, Varma S, Malhotra P, CML with pregnancy: Real challenges in developing nations: Leuk Lymphoma, 2017; 58; 1518-19
17.. Darabi D, Dzik W, Hyperhemolysis syndrome in anemia of chronic disease: Transfusion, 2005; 45; 1930-33
18.. Win N, Yeghen T, Needs M, Use of intravenous immunoglobulin and intravenous methylprednisolone in hyperhaemolysis syndrome in sickle cell disease: Hematology, 2004; 9; 433-36
19.. Petz LD, The sickle cell hemolytic transfusion reaction syndrome: Transfusion, 1997; 37; 382-92
20.. Lubin B, Chiu D, Bastacky J, Abnormalities in membrane phospholipid organization in sickled erythrocytes: J Clin Invest, 1981; 67; 1643-49
21.. Ihanus E, Uotila LM, Toivanen A, Red-cell ICAM-4 is a ligand for the monocyte/macrophage integrin CD11c/CD18: Characterization of the binding sites on ICAM-4: Blood, 2007; 109; 802-10
22.. Win N, Hyperhemolysis syndrome in sickle cell disease: Expert Rev Hematol, 2009; 2; 111-15
23.. Narbey D, Habibi A, Chadebech P, Incidence and predictive score for delayed hemolytic transfusion reaction in adult patients with sickle cell disease: Am J Hematol, 2017; 92; 1340-48
24.. Habibi A, Mekontso-Dessap A, Guillaud C, Delayed hemolytic trans-fusion reaction in adult sickle-cell disease: Presentations, outcomes, and treatments of 99 referral center episodes: Am J Hematol, 2016; 91; 989-94
25.. Aragona E, Kelly MJ, Hyperhemolysis in sickle cell disease: J Pediatr Hematol Oncol, 2014; 36; e54-56
26.. Salama A, Mueller-Eckhardt C, Delayed hemolytic transfusion reactions. Evidence for complement activation involving allogeneic and autologous red cells: Transfusion, 1984; 24; 188-93
27.. Wang RH, Phillips G, Medof ME, Mold C, Activation of the alternative complement pathway by exposure of phosphatidyl ethanolamine and phosphatidyl serine on erythrocytes from sickle cell disease patients: J Clin Invest, 1993; 92; 1326-35
28.. Chadebech P, Habibi A, Nzouakou R, Delayed hemolytic transfusion reaction in sickle cell disease patients: Evidence of an emerging syndrome with suicidal red blood cell death: Transfusion, 2009; 49; 1785-92
29.. Hod EA, Sokol SA, Zimring JC, Spitalnik SL, Hypothesis: Hemolytic transfusion reactions represent an alternative type of anaphylaxis: Int J Clin Exp Pathol, 2009; 2; 71-82
30.. Noizat-Pirenne F, Bachir D, Chadebech P, Rituximab for prevention of delayed hemolytic transfusion reaction in sickle cell disease: Haematologica, 2007; 92; e132-35
31.. Noizat-Pirenne F, Habibi A, Mekontso-Dessap A, The use of rituximab to prevent severe delayed haemolytic transfusion reaction in immunized patients with sickle cell disease: Vox Sanguinis, 2015; 108; 262-67
32.. Win N, Delayed hemolytic transfusion reaction, intravenous immunoglobulin, and rituximab: Transfusion, 2013; 53; 2829-30
33.. Delmonte L, Cantini M, Olivieri O, de Franceschi L, Immunoglobulin-resistant delayed hemolytic transfusion reaction treated with rituximab in an adult sickle cell patient: Transfusion, 2013; 53; 688-89
34.. Littera R, Arras M, Ledda A, Long-term efficacy and tolerance of rituximab for post-transfusional alloimmune haemolytic anaemia in a thalassaemia patient: Br J Haematol, 2007; 140; 114-15
35.. Test ST, Woolworth VS, Defective regulation of complement by the sickle erythrocyte: Evidence for a defect in control of membrane attack complex formation: Blood, 1994; 83; 842-52
36.. Frimat M, Tabarin F, Dimitrov JD, Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome: Blood, 2013; 122; 282-92
37.. Boonyasampant M, Weitz IC, Kay B, Life-threatening delayed hyperhemolytic transfusion reaction in a patient with sickle cell disease: Effective treatment with eculizumab followed by rituximab: Transfusion, 2015; 55; 2398-403
38.. Dumas G, Habibi A, Onimus T, Eculizumab salvage therapy for delayed hemolysis transfusion reaction in sickle cell disease patients: Blood, 2016; 127; 1062-64
39.. Chonat S, Quarmyne MO, Bennett CM, Contribution of alternative complement pathway to delayed hemolytic transfusion reaction in sickle cell disease: Haematologica, 2018; 103(10); e483-85
40.. Vlachaki E, Gavrilaki E, Kafantari K, Successful outcome of hyperhemolysis in sickle cell disease following multiple lines of treatment: The role of complement inhibition: Hemoglobin, 2018; 42(5–6); 339-41
41.. Unnikrishnan A, Pelletier JPR, Bari S, Anti-N and anti-Doa immunoglobulin G alloantibody-mediated delayed hemolytic transfusion reaction with hyperhemolysis in sickle cell disease treated with eculizumab and HBOC-201: Case report and review of the literature: Transfusion, 2019; 59; 1907-10
42.. Mpinganzima C, Haaland A, Holm AGV, Two consecutive episodes of severe delayed hemolytic transfusion reaction in a sickle cell disease patient: Case Rep Hematol, 2020; 2020; 2765012
43.. Weinstock C, Mohle R, Dorn C, Successful use of eculizumab for treatment of an acute hemolytic reaction after ABO-incompatible red blood cell transfusion: Transfusion, 2015; 55; 605-10
44.. Terano C, Ishikura K, Hamada R, Practical issues in using eculizumab for children with atypical haemolytic uraemic syndrome in the acute phase: A review of four patients: Nephrology, 2018; 23; 539-45
45.. Girardi G, Complement inhibition keeps mothers calm and avoids fetal rejection: Immunol Invest, 2008; 37; 645-59
46.. Kelly RJ, Höchsmann B, Szer J, Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria: N Engl J Med, 2015; 373; 1032-39
47.. Kulasekararaj AG, Hill A, Rittinghaus ST, Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study: Blood, 2019; 133; 540-49
48.. Tomazos I, Sierra JR, Johnston KM, Cost burden of breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria receiving ravulizumab versus eculizumab: Hematology, 2020; 25; 327-34
49.. Brodsky RA, Young NS, Antonioli E, Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria: Blood, 2008; 111; 1840-47
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952577
Most Viewed Current Articles
07 Dec 2021 : Case report  22,759,844
22,759,844
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  176,022
176,022
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,540
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,552
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133