17 October 2021: Articles 
Spontaneous Recovery of Hemophagocytic Lymphohistiocytosis Due to Primary Epstein-Barr Virus Infection in an Adult Patient
Unusual clinical course
Yuichiro Matsuo1ADEF, Keiichi Iwanami2ADEF*, Eiji Hiraoka1ADEF, Rentaro Oda3DEDOI: 10.12659/AJCR.933272
Am J Case Rep 2021; 22:e933272






Abstract
BACKGROUND: Hemophagocytic lymphohistiocytosis (HLH) is a rare life-threatening systemic hyperinflammatory condition. Most adult HLH cases are secondary to infection, malignancy, and rheumatic diseases. Epstein-Barr virus (EBV) infection is the most frequent cause of infection-induced HLH. Early treatment with dexamethasone, etoposide, and cyclosporine is generally recommended for adult patients with HLH. However, this treatment regimen was established based on pediatric clinical trial data; thus, its efficacy and validity in adults remain unclear. Because little is known about the disease course of untreated adult EBV-associated HLH (EBV-HLH), we report a case of an adult patient who recovered from EBV-HLH spontaneously without specific treatment and discuss potential treatment strategies.
CASE REPORT: A 34-year-old man presented to the emergency department with a 7-day history of fever, headache, and sore throat. The main laboratory test abnormalities were elevated liver enzymes, hyperbilirubinemia, hypertriglyceridemia, and hyperferritinemia. Serologic tests confirmed acute primary EBV infection. He was diagnosed with EBV-HLH based on the HLH-2004 diagnostic criteria and the HLH probability calculator (HScore). Because he was clinically stable, we did not initiate immunosuppressive/cytotoxic treatment targeting HLH. High-grade fever persisted, but the abnormalities in his laboratory data improved spontaneously, and he did not develop major organ failure. His fever resolved on day 29 without HLH-specific treatment.
CONCLUSIONS: In clinically stable adult patients with EBV-HLH without major organ failure, it might be an acceptable alternative to observe the patient for several weeks before initiating HLH-specific treatment. Further research is required to better predict the subset of patients who can be safely observed without treatment.
Keywords: Epstein-Barr Virus Infections, Immunosuppressive Agents, Lymphohistiocytosis, Hemophagocytic, Child, Cyclosporine, Etoposide, Herpesvirus 4, Human, Humans, Male
Background
Hemophagocytic lymphohistiocytosis (HLH) is a rare life-threatening systemic hyperinflammatory condition that is caused by primary immune deficiency (primary HLH) or an acquired failure of immune response (secondary HLH) [1]. It has been hypothesized that the impaired functioning of CD8+ T cells or natural killer (NK) cells and termination of immune response result in hypercytokinemia and uncontrolled activation of macrophages, leading to the hyperinflammatory state of HLH [2]. Common symptoms of HLH are fever and rash, and typical laboratory findings are pancytopenia, elevated liver enzyme levels, elevated D-dimer, prolonged prothrombin time and partial thromboplastin time, hypertriglyceridemia, hyperferritinemia, and coagulation abnormalities, such as hypofibrinogenemia [3].
In most adult cases, HLH is secondary to infection, malignancy, and rheumatic diseases. Epstein-Barr virus (EBV) infection is the most frequent cause of infection-induced HLH [4]. EBV primarily infects B cells, but T cells are also infected in some individuals, including immunocompetent adults. The infected CD8+ T cells lose their function, thereby leading to the development of HLH [5].
The diagnosis of HLH is established when there is a molecular diagnosis consistent with HLH, or when more than 5 out of 8 of the HLH-2004 diagnostic criteria are met: (1) fever, (2) splenomegaly, (3) Cytopenia affecting more than 2 of 3 cell lineages, (4) hypertriglyceridemia and/or hypofibrinogenemia, (5) hemophagocytosis in bone marrow, spleen, or lymph nodes, (6) low or absent NK cell activity, (7) hyperferritinemia, and (8) elevated levels of soluble interleukin (IL)-2 receptor [6]. Timely diagnosis is essential since HLH is associated with high mortality [7], and a delay in diagnosis can lead to poor outcome [1].
Once HLH is confirmed, early treatment based on regimens from the HLH-94 clinical trial [8] or the HLH-2004 clinical trial [6] is generally recommended. Both clinical trial-based regimens consist of combined immunosuppressive/cytotoxic treatment with dexamethasone, etoposide, and cyclosporine. However, these treatment regimens were established from pediatric clinical trial data [9], and, thus, their efficacy in adults remains unclear. Moreover, adverse effects related to these treatments are a major concern. Recently, an expert opinion statement from a working group on adult HLH was published [10]. The authors suggest a more conservative approach with glucocorticoid alone as initial treatment in patients with non-severe EBV-HLH. However, this statement also lacks underlying clinical evidence.
Little is known about the natural disease course of untreated EBV-HLH in adult patients. Here, we report a case of an adult patient who recovered from HLH due to primary EBV infection spontaneously without specific treatment and discuss potential treatment strategies. This is the third reported case of spontaneous recovery of EBV-HLH in an adult patient [11,12] and will advance medical knowledge about a condition regarding which evidence is lacking.
Case Report
A 34-year-old man presented to the emergency department with a 7-day history of fever, headache, and sore throat. On the day of symptom onset (day 0), a week prior, he sought care at a clinic, where he was prescribed a 7-day course of amoxicillin/clavulanic acid and levofloxacin. However, his symptoms persisted. His medical history was unremarkable. On admission (day 7), his temperature, blood pressure, and heart rate were 38.4°C, 125/86 mm Hg, and 97 beats/min, respectively. Physical examination was notable for conjunctival icterus, swollen tonsils covered with slough, tender bilateral anterior cervical lymphadenopathy, and hepatomegaly. Splenomegaly was undetectable from physical examination. Laboratory tests on admission showed lymphocytosis with 1.0% of atypical lymphocytes, thrombocytopenia, elevated liver enzyme levels, hyperbilirubinemia, prolonged prothrombin time and activated partial thromboplastin time, and hypertriglyceridemia. Hyperferritinemia and elevated levels of soluble IL-2 receptor were also noted in additional tests (Table 1). NK cell activity was not measured. Computed tomography of the chest and abdomen revealed hepatosplenomegaly. EBV antibody titers were positive for viral capsid antigen (VCA)-IgM and VCA-IgG and negative for Epstein-Barr nuclear antigen, thereby confirming primary EBV infection. Serologic tests for cytomegalovirus (CMV) and human immunodeficiency virus were negative. His clinical features and laboratory data were consistent with infectious mononucleosis due to primary EBV infection.
Given the presence of fever, splenomegaly, hypertriglyceridemia, hyperferritinemia, and elevated level of soluble IL-2 receptor, he also met the HLH-2004 diagnostic criteria [6]. He also had thrombocytopenia; however, the platelet count was slightly above the cut-off value of the diagnostic criteria. He did not have neutropenia or anemia. These diagnostic criteria, however, were developed for children, and have not been validated for adults. Therefore, we also referred to the HLH probability calculator (HScore), which is for adult patients with HLH [13]; the calculated score was 200, indicating an 80% to 88% probability of HLH. Along with the presence of acute EBV infection, we diagnosed him with EBV-HLH. However, we decided not to administer the combination of immunosuppressants and cytotoxic agents recommended in the HLH-94 or HLH-2004 protocol since he was clinically stable, without major organ failure or impending severe cytopenia. Instead, he received supportive care.
Thrombocytopenia and prolonged prothrombin time and activated partial thromboplastin time improved by day 12, but he continued to have high-grade fever, elevated liver enzyme levels, and hyperbilirubinemia. On day 22 of symptoms, he developed a macular rash on his trunk and extremities (Figure 1). The size of the rash fluctuated in a manner that corresponded to the spikes in fever, which was similar to the “salmon-pink” rash of adult-onset Still’s disease (AOSD). However, the diagnosis of AOSD could not be established; exclusion of other inflammatory diseases is necessary for the diagnosis of AOSD, and EBV-HLH matched his clinical course best. Therefore, we regarded this rash as a skin manifestation of HLH, and withheld initiation of immunosuppressive therapy targeting AOSD.
Bone marrow examination on day 24 ruled out underlying hematological malignancies and revealed a normocellular marrow, without an increase in blasts or dysplasia. Hemophagocytosis was absent, but epithelioid cell granulomas were present in bone marrow aspirate clot samples (Figure 2). The presence of epithelioid cell granulomas in bone marrow samples was suggestive of macrophage activation. The presence of epithelioid cell granulomas also raised suspicion for
He did not have severe cytopenia or major organ failure, and his liver enzyme levels and hyperferritinemia were gradually improving; therefore, he was discharged from the hospital. On day 29, upon his follow-up at the outpatient clinic, his fever had resolved, and his rash had disappeared (Figure 3). He was monitored at the outpatient clinic for the next 4 months, and he remained afebrile and without clinical symptoms.
Discussion
We observed a case of EBV-HLH in an adult patient who recovered spontaneously without specific treatment. Upon initial presentation, there remained the possibility that his clinical features and laboratory abnormalities could be explained solely by infectious mononucleosis caused by primary EBV infection. However, there were several aspects which were inconsistent with simple infectious mononucleosis. First, in most cases of infectious mononucleosis, fever resolves within 14 days [17], but in our patient fever lasted more than 4 weeks. Second, mild hepatic involvement with elevated liver enzyme levels (2–3 times the upper limit of normal) is common in infectious mononucleosis, and it has been reported that a minority of patients present with jaundice [18,19]. Extreme hyperferritinemia over 10 000 ng/mL can be seen in not only HLH but also in acute hepatitis [20,21]. In our patient, however, even after liver enzyme levels approached normal levels, fever did not abate, and transient macular rash developed, which indicated that he developed HLH rather than mere EBV-hepatitis. Finally, the fact that our patient fulfilled the HLH-2004 criteria and exhibited a sufficient HScore strongly supported the probability of HLH. If the EBV-related symptoms had lasted for more than 3 months, a diagnosis of chronic active EBV disease would have been made [22,23], but our patient achieved defervescence on day 29. Thus, we diagnosed our patient with EBV-HLH.
Our clinical dilemma was whether to initiate combined immunosuppressive/cytotoxic treatment with dexamethasone, etoposide, and cyclosporine based on the HLH-94 and the HLH-2004 clinical trials, since it is reported that the mortality rate of EBV-HLH is extremely high [24–26].
In pediatric patients with HLH, the introduction of HLH-94-based treatment has led to a dramatic improvement in prognosis. The estimated 5-year probability of survival has improved to 54% after its introduction [9]; prior to that, HLH had been almost always fatal [27]. However, no prospective clinical trials have been performed to evaluate the efficacy of HLH-94-based treatment or HLH-2004-based treatment in adult patients with HLH.
Some other novel treatment strategies are being evaluated. The efficacy of emapalumab, a human anti-interferon-γantibody, has shown positive results in a single-arm trial of pediatric primary HLH patients [28]. A trial of emapalumab is also ongoing in adult HLH patients (NCT03985423). Ruxolitinib, a JAK inhibitor, was also suggested to be effective in a single-arm pilot trial of secondary HLH in adults [29]. A larger trial evaluating the efficacy of ruxolitinib in HLH patients, including both children and adults who were refractory to HLH-94-based treatment, is now ongoing (NCT04120090). However, the completed and planned clinical trials of these agents are all single-arm trials, and it would still be difficult to guide an evidence-based treatment strategy in adult HLH patients.
The reported overall mortality of adult patients with HLH is high, varying from approximately 40% to 70% [7,26,30–32]. The reported mortality of adult patients with EBV-HLH is also high, varying from 75% to 95% [24–26]. The majority of the patients included in these adult EBV-HLH studies have received intensive immunosuppressive/cytotoxic treatment, including HLH-94 and HLH-2004 clinical trial-based regimens, and little is known about the natural course and prognosis of adult EBV-HLH without treatment.
In addition to the lack of evidence on treating adult EBV-HLH cases with the clinical trial-based regimens, adverse effects related to immunosuppressive/cytotoxic treatment are a major concern. The risk of infection increases with the use of either drug regimen from the HLH clinical trials [33–35], and etopo-side is associated with hematologic toxicity [36] and secondary hematologic malignancies [37]. Moreover, from pathophysio-logical aspects, the possibility remains that immunosuppressive therapy can worsen the condition of EBV infection and EBV-HLH, since cell-mediated immunity plays a central role in eradicating EBV [17]. Therefore, immunosuppressive therapy can impair a patient’s ability to eliminate EBV infection, leading to severe infections and complications, including the development and exacerbation of HLH. For example, patients with inflammatory bowel disease undergoing thiopurine therapy carry an increased risk for virus-induced HLH, the majority of which are due to primary EBV and CMV infections [38]. Several cases of EBV-HLH due to reactivation of EBV during immunosuppressive treatment for rheumatologic diseases have also been reported [39,40]. Although these reports do not directly indicate that immunosuppressive therapy is uniformly harmful to patients with EBV infection, we need to be aware of the potential harms of introducing immunosuppressive treatment to patients with EBV-HLH.
Our literature review revealed that only 2 other cases of spontaneous resolution of EBV-HLH have been reported in adults to date [11,12]. The first case was a 19-year-old man who presented with 3 days of fever and abdominal pain. During his clinical course, he developed pancytopenia (nadir blood cell count: white blood cells, 900/μL; hemoglobin, 9.8 g/dL; and platelets, 69 000/μL), hyperferritinemia (2016 ng/mL), and elevated levels of soluble IL-2 receptor (2518 IU/mL). The authors considered initiating treatment with the HLH-2004 clinical trial-based regimen, but the patient was observed without treatment instead because his clinical condition improved and blood cell counts recovered over the first 2 weeks. His blood cell counts normalized after 1 month from initial presentation [11]. The second case was a 23-year-old woman who presented with fever and fatigue. She had pancytopenia (white blood cells, 1500/μL; hemoglobin, 8 g/dL; and platelets, 59 000/μL), elevated liver enzymes (aspartate aminotransferase, 593 IU/L; alanine aminotransferase, 1321 IU/L; and lactate dehydrogenase, >2000 IU/L), hyperferritinemia (7665 ng/mL), and elevated levels of soluble IL-2 receptor (2458 IU/mL). By the time her diagnosis of EBV-HLH was confirmed, she was already exhibiting gradual clinical improvement; her fever and laboratory data abnormalities improved over a period of 2 to 3 weeks without treatment [12]. In both cases, the patients did not develop severe cytopenia or show any signs of major organ failure, and spontaneous clinical improvement was observed during the next several weeks. The authors of the former case report suggested observing patients for 4 weeks without initiating treatment if the patients remain clinically stable and without any life-threatening conditions [11].
Considering the uncertainty of immunosuppressive/cytotoxic treatment efficacy in adult patients with EBV-HLH and the potential harms, it may be reasonable to observe low-risk patients, without initiating treatment. Unfortunately, there is no generally accepted risk stratification strategy for adult patients with HLH. In a French cohort of 162 adult patients with HLH, advanced age (median age of non-survivors was 56 years), lower platelet count (median platelet count of non-survivors was 40 600/μL), underlying lymphoma, and initial treatment without etoposide were features associated with poor short-term prognosis [7]. In another cohort from China that included 174 adult patients with HLH, low lymphocyte count (<500/μL) and hypofibrinogenemia (<150 mg/dL) were associated with poor long-term prognosis [31]. Notably, underlying viral infection was not a factor indicating poor prognosis. In another recent cohort from China that evaluated 126 adult patients with HLH without malignancy, age over 45 years, platelet count below 35 000/μL, serum ferritin over 20 000 ng/mL, and a diagnosis of EBV-HLH were associated with poor long-term prognosis [26].
Fewer data are available on the prognostic factors of adult patients with EBV-HLH. In a single-center study of 23 adult patients with EBV-HLH in China, bone marrow suppression, poor performance status, high lactate dehydrogenase level (cut-off level 1172 IU/L), and treatment not based on the HLH-2004 clinical trial regimens were associated with poor prognosis [24]. However, we need to consider that the majority of patients included in these studies underwent some kind of immunosuppressive/cytotoxic therapy, including HLH-94/HLH-2004 clinical trial-based regimens, and do not reflect the outcome of adult patients with HLH who do not receive treatment. It is noteworthy, however, that treatment with non-HLH clinical trial-based regimens and non-etoposide-containing regimens have been associated with poor prognosis in some studies.
Considering the high mortality of adult patients with EBVHLH, initiation of either HLH-94/HLH-2004 clinical trial-based regimens without delay is crucial in patients with severe HLH with major organ failure (eg, central nervous system involvement, hepatic failure) or deteriorating clinical status. In clinically stable patients without major organ failure and without known clinical factors associated with poor prognosis (severe cytopenia, hypofibrinogenemia, or marked hyperferritinemia), it may be reasonable to observe the patient for up to 4 weeks before initiating HLH-specific treatment.
Conclusions
We observed a case of EBV-HLH in an adult patient who spontaneously recovered without specific treatment. It might be an acceptable alternative to observe a patient for several weeks without initiating specific treatment if the patient is clinically stable and does not exhibit any signs of major organ failure. Clinical evidence regarding the prognosis and treatment of adult patients with HLH is lacking, and further research is required to better predict the subset of patients who can be safely observed without treatment.
Figures
Tables
Table 1.. Laboratory data.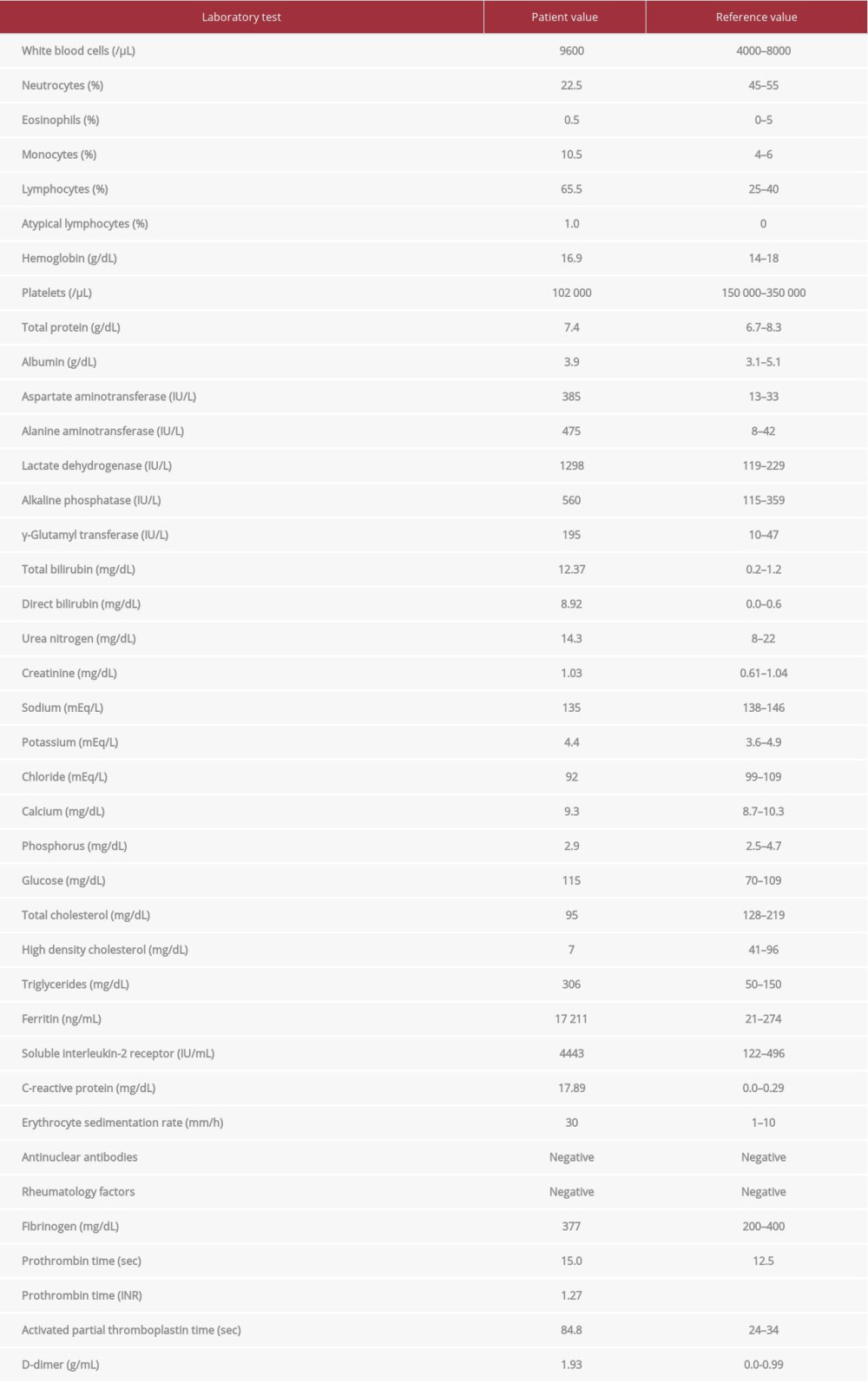
References:
1.. Al-Samkari H, Berliner N, Hemophagocytic lymphohistiocytosis: Annu Rev Pathol, 2018; 13; 27-49
2.. Usmani GN, Woda BA, Newburger PE, Advances in understanding the pathogenesis of HLH: Br J Haematol, 2013; 161; 609-22
3.. Griffin G, Shenoi S, Hughes GC, Hemophagocytic lymphohistiocytosis: An update on pathogenesis, diagnosis, and therapy: Best Pract Res Clin Rheumatol, 2020; 34; 101515
4.. Maakaroun NR, Moanna A, Jacob JT, Albrecht H, Viral infections associated with haemophagocytic syndrome: Rev Med Virol, 2010; 20; 93-105
5.. Kawaguchi H, Miyashita T, Herbst H, Epstein-Barr virus-infected T lymphocytes in Epstein-Barr virus-associated hemophagocytic syndrome: J Clin Invest, 1993; 92; 1444-50
6.. Henter J-I, Horne A, Aricó M, HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis: Pediatr Blood Cancer, 2007; 48; 124-31
7.. Arca M, Fardet L, Galicier L, Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: Impact of triggering disease and early treatment with etoposide: Br J Haematol, 2015; 168; 63-68
8.. Henter J-I, Samuelsson-Horne A, Aricò M, Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation: Blood, 2002; 100; 2367-73
9.. Trottestam H, Horne A, Aricò M, Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: Long-term results of the HLH-94 treatment protocol: Blood, 2011; 118; 4577-84
10.. La Rosée P, Horne A, Hines M, Recommendations for the management of hemophagocytic lymphohistiocytosis in adults: Blood, 2019; 133; 2465-77
11.. Belyea B, Hinson A, Moran C, Spontaneous resolution of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis: Pediatr Blood Cancer, 2010; 55; 754-56
12.. Chahine Z, Jayakrishnan T, Samhouri Y, Fazal S, Haemophagocytic lymphohistiocytosis that spontaneously resolved: A case of EBV: BMJ Case Rep, 2020; 13; e235544
13.. Fardet L, Galicier L, Lambotte O, Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome: Arthritis Rheumatol, 2014; 66; 2613-20
14.. McCall CM, Mudali S, Arceci RJ, Flow cytometric findings in hemophagocytic lymphohistiocytosis: Am J Clin Pathol, 2012; 137; 786-94
15.. Goel S, Polski JM, Imran H, Sensitivity and specificity of bone marrow hemophagocytosis in hemophagocytic lymphohistiocytosis: Ann Clin Lab Sci, 2012; 42; 21-25
16.. Ho C, Yao X, Tian L, Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability: Am J Clin Pathol, 2014; 141; 62-71
17.. Johannsen EC, Kaye KM, Epstein-Barr virus (infectious mononucleosis, Epstein-Barr virus – associated malignant diseases, and other diseases): Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 2019; 1872-90, Philadelphia, Elsevier
18.. Kofteridis DP, Koulentaki M, Valachis A, Epstein Barr virus hepatitis: Eur J Intern Med, 2011; 22; 73-76
19.. Schechter S, Lamps L, Epstein-Barr virus hepatitis: A review of clinicopathologic features and differential diagnosis: Arch Pathol Lab Med, 2018; 142; 1191-95
20.. Kotoh K, Ueda A, Tanaka M, A high prevalence of extreme hyperferritinemia in acute hepatitis patients: Hepat Med, 2009; 1; 1-8
21.. Otrock ZK, Hock KG, Riley SB, Elevated serum ferritin is not specific for hemophagocytic lymphohistiocytosis: Ann Hematol, 2017; 96; 1667-72
22.. Kimura H, Cohen JI, Chronic active Epstein-Barr virus disease: Front Immunol, 2017; 8; 1867
23.. Cohen JI, Jaffe ES, Dale JK, Characterization and treatment of chronic active Epstein-Barr virus disease: A 28-year experience in the United States: Blood, 2011; 117; 5835-49
24.. Shao X, Xu Y, Xu X, Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in adults: A retrospective analysis of 23 patients in China: Isr Med Assoc J, 2018; 20; 80-85
25.. Lai W, Wang Y, Wang J, Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in adults and adolescents-a life-threatening disease: Analysis of 133 cases from a single center: Hematology, 2018; 23; 810-16
26.. Yoon J-H, Park S-S, Jeon Y-W, Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy: Haematologica, 2019; 104; 269-76
27.. Janka GE, Familial hemophagocytic lymphohistiocytosis: Eur J Pediatr, 1983; 140; 221-30
28.. Locatelli F, Jordan MB, Allen C, Emapalumab in children with primary hemophagocytic lymphohistiocytosis: N Engl J Med, 2020; 382; 1811-22
29.. Ahmed A, Merrill SA, Alsawah F, Ruxolitinib in adult patients with secondary haemophagocytic lymphohistiocytosis: An open-label, single-centre, pilot trial: Lancet Haematol, 2019; 6; e630-37
30.. Parikh SA, Kapoor P, Letendre L, Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis: Mayo Clin Proc, 2014; 89; 484-92
31.. Zhang Q, Li L, Zhu L, Adult onset haemophagocytic lymphohistiocytosis prognosis is affected by underlying disease: Analysis of a single-institution series of 174 patients: Swiss Med Wkly, 2018; 148; w14641
32.. Rivière S, Galicier L, Coppo P, Reactive hemophagocytic syndrome in adults: A retrospective analysis of 162 patients: Am J Med, 2014; 127; 1118-25
33.. Youssef J, Novosad SA, Winthrop KL, Infection risk and safety of corticosteroid use: Rheum Dis Clin North Am, 2016; 42; 157-76 , ix–x
34.. Singh N, Infectious complications in organ transplant recipients with the use of calcineurin-inhibitor agent-based immunosuppressive regimens: Curr Opin Infect Dis, 2005; 18; 342-45
35.. Bow EJ, Infection risk and cancer chemotherapy: The impact of the chemo-therapeutic regimen in patients with lymphoma and solid tissue malignancies: J Antimicrob Chemother, 1998; 41(Suppl. D); 1-5
36.. Kaul S, Srinivas NR, Igwemezie LN, Barbhaiya RH, A pharmacodynamic evaluation of hematologic toxicity observed with etoposide phosphate in the treatment of cancer patients: Semin Oncol, 1996; 23; 15-22
37.. Ezoe S, Secondary leukemia associated with the anti-cancer agent, etoposide, a topoisomerase II inhibitor: Int J Environ Res Public Health, 2012; 9; 2444-53
38.. Li Y, Li CF, Zhang J, Features of patients with inflammatory bowel diseases who develop hemophagocytic lymphohistiocytosis: Int J Colorectal Dis, 2016; 31; 1375-76
39.. Kawashiri S, Nakamura H, Kawakami A, Emergence of Epstein-Barr virus-associated haemophagocytic syndrome upon treatment of systemic lupus erythematosus: Lupus, 2006; 15; 51-53
40.. Hayakawa I, Shirasaki F, Ikeda H, Reactive hemophagocytic syndrome in a patient with polyarteritis nodosa associated with Epstein-Barr virus reactivation: Rheumatol Int, 2006; 26; 573-76
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133