20 January 2022: Articles 
A Missed Case of Area Postrema Syndrome Presenting with Neuromyelitis Optica Spectrum Disorder
Mistake in diagnosis, Rare disease
Faisal Khan1ABCDEF*, Neha Sharma2ABCDEF, Moin Ud Din2ABCDEF, Munmun Aziz3ABCDEFDOI: 10.12659/AJCR.934649
Am J Case Rep 2022; 23:e934649






Abstract
BACKGROUND: Neuromyelitis optica spectrum disorder (NMOSD), which is also known as Devic disease, is a chronic disorder of the brain and spinal cord that includes inflammation of the optic nerve and spinal cord. Area postrema syndrome (APS) is due to involvement of the bulbar emetic reflex center, and has previously been described in NMOSD. Patients with APS may present with nausea, vomiting, or hiccups. This report is of a 33-year-old Asian American woman with history of APS who presented with NMOSD.
CASE REPORT: A 33-year-old Southeast Asian woman, 2 months postpartum, presented with fever, hypersomnolence, altered mental status, and difficulty ambulating. Neurological examination revealed a lethargic woman with poor attention span, broad-based gait ataxia, and positive Romberg’s sign. Laboratory work-up showed sodium 123 milliequivalent/L (mEq/L). Brain magnetic resonance imaging (MRI) with contrast revealed bilateral, non-enhancing, patchy fluid-attenuated inversion recovery (FLAIR) hyperintensities in the anteroinferomedial thalamus extending to the mammillary bodies. Additional history revealed hospitalization for intractable nausea, vomiting, and hiccups 2 years ago. NMOSD was confirmed with positive AQP-4 antibody, prompting treatment with intravenous (i.v.) methylprednisolone, followed by plasmapheresis. Repeat brain MRI showed mild improvement of bilateral thalamic FLAIR hyperintensities and no clinical recurrence was reported with Rituximab treatment.
CONCLUSIONS: This case highlights the importance of the diagnostic diligence required for NMOSD diagnosis. Multiple etiologies can mimic the clinical presentation of acute diencephalic syndrome; thus, a broad differential needs to be considered. This report presents the diagnostic work-up and management of a patient with a complex neurological condition that was diagnosed as NMOSD.
Keywords: Aquaporin 4, Area Postrema, Diencephalon, Neuromyelitis Optica, Wernicke Encephalopathy, Adult, Female, Humans, Magnetic Resonance Imaging, Nausea, Vomiting
Background
Neuromyelitis optica spectrum disorder has significant diagnostic complexity due to its myriad of clinical presentations. The spectrum of core clinical characteristics includes optic neuritis, longitudinally extensive transverse myelitis, area postrema syndrome, acute cerebral syndrome, acute brainstem syndrome, and acute diencephalic syndrome [1]. Each of these characteristics can be caused by a plethora of conditions; thus, a broad differential needs to be considered.
Prompt initiation of long-term immunosuppression is imperative to prevent progression and recurrence. Consequences of untreated NMOSD are dire, as 50% of cases develop residual deficits with chronic disability, while the mortality rate is 33% within 5 years of developing initial symptoms [2].
This report is of a 33-year-old Asian American woman with a history of APS who presented with NMOSD.
Case Report
A 33-year-old Southeast Asian woman, 2 months postpartum, actively breast-feeding, with a past medical history of chronic episodic migraines, experienced fever and sweats. Testing for severe acute respiratory syndrome coronavirus 2 (SARSCoV-2) via polymerase chain reaction (PCR) and antibody was negative. The patient presented to the Emergency Department (ED) with a 7-day history of severe hypersomnolence, altered mental status, and difficulty walking. Her husband reported she had tangential thought patterns and flight of ideas. Four weeks previously, the patient went to her primary care physician, reporting nausea and diaphoresis, with a fever of 38.6°C. She had an abnormal urinalysis result, for which ciprofloxacin was initiated due to a suspected urinary tract infection (UTI) and she was also advised to adequately hydrate. She denied any current headaches, fever, and dizziness. She also denied a history of alcohol, smoking, and illicit drug use. Additionally, the patient denied recent vaccination or infection. Vital signs taken on arrival were blood pressure 123/92 millimeters of mercury (mmHg), heart rate 98 beats per minute, respiratory rate 18 breaths per minute, temperature 37.1°C, and SpO2 98% on room air. Cardiopulmonary examination results were within normal limits. A neurological examination revealed a lethargic woman with limited attention span, but normal speech and comprehension. Her Epworth scale was 17, and her Glasgow coma scale showed motor response (6), visual response (4), and verbal response (4). Cranial nerve examination showed pupils were equal, round, and reactive, extraocular muscle movements were intact, and face and tongue showed no asymmetry. No focal motor deficits were noted, and deep tendon reflexes were symmetric and intact. No cortical or peripheral sensory deficits were noted. Coordination testing showed intact finger-to-nose, but heel-to-shin testing was performed with difficulty. Romberg testing was positive. Gait analysis revealed broad-based ataxia, requiring 1-person assist. Complete blood count and comprehensive metabolic panel was within normal limits, except for sodium 123 mEq/L. Non-contrast brain computed tomography (CT) showed no signs of acute infarct, hemorrhage, or other intracranial abnormalities.
The patient was admitted and subsequent neuroimaging with brain MRI with contrast revealed bilateral, non-enhancing, patchy FLAIR hyperintensities in the anteroinferomedial thalamus extending to the mammillary bodies (Figure 1). A provisional diagnosis of non-alcoholic Wernicke’s encephalopathy was made and the patient was empirically started on intravenous (i.v.) thiamine 500 milligrams (mg) 3 times daily. Her pre-infusion thiamine level was reported as 158 nanomoles/liter (nmol/L). Despite post-infusion thiamine levels reaching 1200 nmol/L, the patient remained symptomatic. Brain and neck magnetic resonance angiography without contrast showed no evidence of stenosis in the extra- or intracranial circulation. Brain magnetic resonance venography without contrast showed no thrombosis or hemodynamically significant stenosis, apart from a small left transverse sinus suspected to be a normal anatomical variant.
Electroencephalogram revealed mild diffuse slowing (7 Hertz) with no epileptiform discharges, intermixed with disorganized and poorly sustained sleep architecture. Hypercoagulable panel revealed low activated partial thromboplastin time (22.4 s) and high protein C (177). Autoimmune panel, human immunodeficiency virus, and
On re-interrogation of the patient, she revealed she had been hospitalized 2 years ago for intractable nausea, vomiting, and hiccups that lasted for 3 weeks. Esophagogastroduodenoscopy at that time revealed mild gastritis. Brain MRI with and without contrast revealed no acute ischemia, hemorrhage, or hydrocephalus. The symptoms self-resolved with no recurrence, and the underlying etiology remained undetermined. Based on this additional information, the suspicion of NMOSD was raised, for which serological work-up for AQP-4 antibodies was initiated. The AQP-4 immunoglobulin G (IgG) antibody test result came back positive and myelin oligodendrocyte glycoprotein (MOG) antibody was negative. Her current symptomatology was consistent with acute diencephalic syndrome, a subset of NMOSD. A 5-day course of high-dose i.v. methylprednisolone sodium succinate treatment was initiated and modafinil was switched to armodafinil 250 mg once in the morning for persistent hypersomnolence. The patient was given 5 sessions of plasmapheresis, which led to improvement in her altered mental status and ataxia, but she remained symptomatic with hypersomnolence. After a discussion with the patient and family regarding risks and benefits, and due to health insurance limitations, she was started on rituximab. At 3-month follow-up, the patient reported no clinical flare-up despite remaining symptomatic with hypersomnia. A repeat brain MRI with and without contrast showed mild improvement of the bilateral thalamic FLAIR hyperintensities.
Discussion
Here we discuss an interesting and complex case of a 33-year-old, Southeast Asian woman, 2 months postpartum, who presented with fever, hypersomnolence, altered mental status, and difficulty ambulating. We will be addressing the challenges faced in the diagnosis and management of neuromyelitis optica spectrum disorder.
Neuromyelitis optica spectrum disorder continues to mesmerize physicians across various disciplines due to overlapping clinical manifestations. The diagnostic criteria for NMOSD have evolved over the last 2 decades [1]. The latest criteria, proposed by Wingerchuk et al in 2015, has increased NMOSD diagnostic sensitivity by 76% by incorporating diverse clinical manifestations and radiological findings along with the AQP-4 antibody status [1,3].
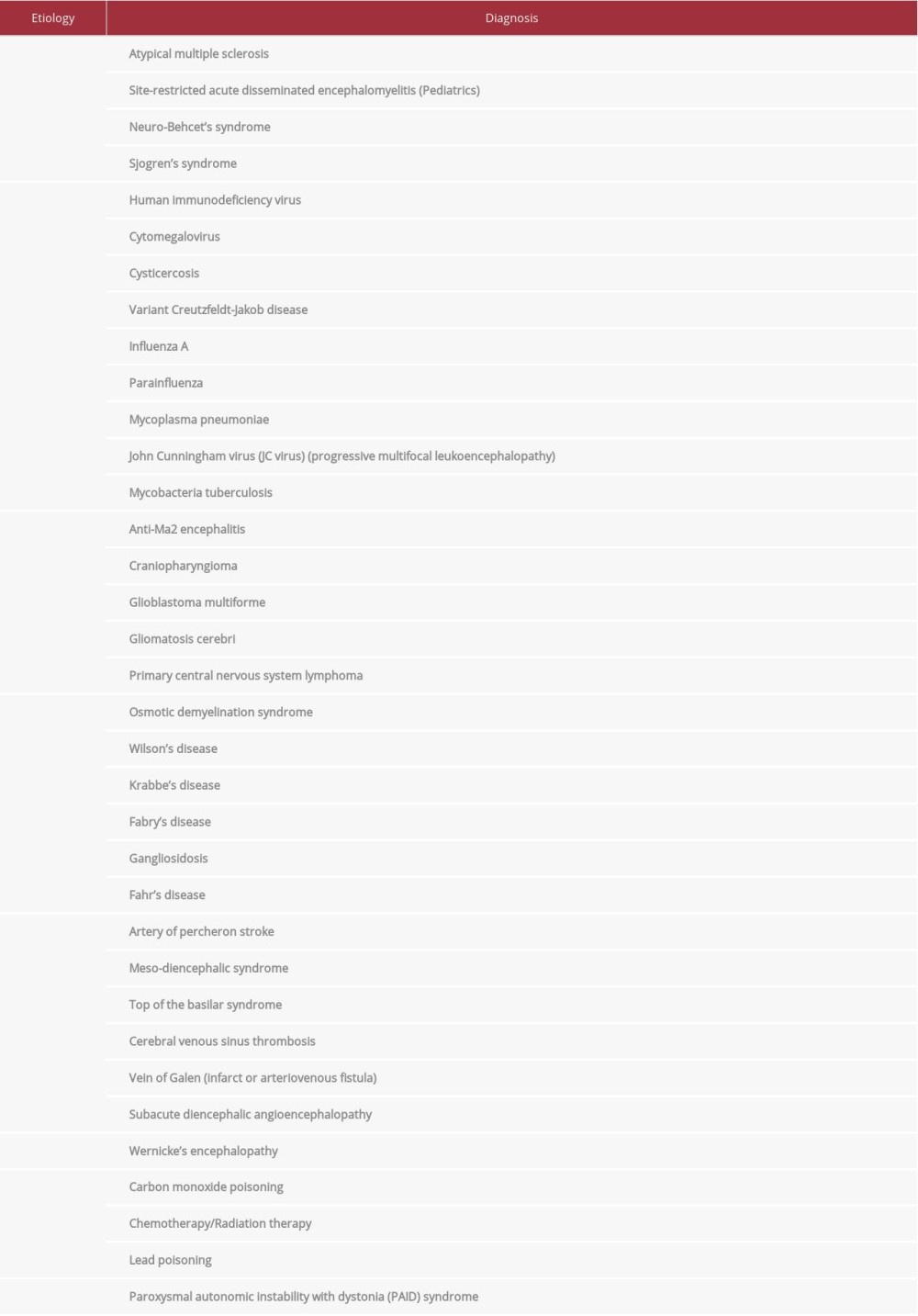
Although our patient presented with features consistent with acute diencephalic syndrome, etiological diagnosis remained a challenge due to broad differentials (Table 1). Acute diencephalic syndrome can present with varying combinations of symptoms including hypersomnia, ataxia, anorexia, inappropriate diuresis, temperature dysregulation, and endocrinological abnormalities, including secondary amenorrhea, galactorrhea, hyperprolactinemia, and hypothyroidism [4–6].
Excessive daytime sleepiness was the main presenting feature of our patient. Hypersomnia in NMOSD can present as both an isolated symptom or as part of acute diencephalic syndrome [7]. The pathogenesis involves damage to orexin neurons along the central projections secondary to hypothalamic lesions, and is usually associated with low CSF orexin levels [7]. Pathological hypersomnia can be extremely disabling and refractory to treatment, as seen in our patient.
Another disabling symptom in our patient was broad-based gait ataxia, requiring 1-person assist. A neurological examination revealed positive Romberg sign and difficulty in performing heel-to-shin, but she had intact finger-to-nose testing and was absent specific cerebellar signs including scanning speech, nystagmus, and dysdiadochokinesia. Sensory ataxia can result from pathologies along the neuraxis at multiple levels. The thalamus is vulnerable to insults from multiple etiologies and, being the main sensory relay station, this can cause severe sensory gait ataxia [8].
Our patient’s vital signs revealed a maximum temperature of 38.6°C, which was initially attributed to her urinary tract infection. Temperature dysregulation, as a symptom of acute diencephalic syndrome, can occur in NMOSD [9]. Inflammation around the preoptic nucleus of the hypothalamus, mediated by the AQP-4 antibody complex, can raise the set-point for core body temperature [9]. Fever of unknown origin with or without other hypothalamic symptoms, especially in young women, should be investigated for NMOSD [9].
Our patient presented with moderate to severe hyponatremia (123 mEq/L), which was initially thought to be associated with psychogenic polydipsia. Significant hyponatremia has been well described in patients with NMOSD, both during the initial attack, as well during relapse, with 60.8% presenting with levels less than 125 mEq/L [10]. The pathogenesis of the hyponatremia is either dilutional via syndrome of inappropriate diuretic hormone secretion, or depletional via cerebral salt wasting syndrome [10]. These etiologies should be differentiated promptly to initiate appropriate treatment such as fluid restriction or resuscitation [10]. Our patient’s hyponatremia responded to water restriction and she was discharged on salt tablets.
Wernicke’s encephalopathy was the initial diagnosis considered due to our patient’s clinical and neuroradiological presentation. Our patient denied alcohol use, but did fulfill 2 of the 4 Caine’s criteria, with findings of ataxia and altered mental status [11]. Our patient’s brain MRI was reviewed by 2 independent American board-certified neuroradiologists, who raised the possibility of Wernicke’s encephalopathy as the primary diagnosis. Our group has previously reported isolated “pulvinar/ hockey stick sign” in patients with non-alcoholic Wernicke’s encephalopathy [8]. Our patient did not respond to thiamine supplementation. Additionally, the pre-infusion thiamine levels available on day 4 of hospitalization were normal, refuting the possibility of non-alcoholic Wernicke’s encephalopathy.
Cerebral venous thrombosis (CVT) was another possible differential in our patient due to her potential hypercoagulable state and abnormal MRI FLAIR findings of thalamic hyperintensities. However, the absence of thrombosis and normal venous anatomy with congenital variation on magnetic resonance venography ruled out CVT in our patient.
Other differential diagnoses that were considered included site-restricted acute disseminated encephalomyelitis, myelin oligodendrocyte glycoprotein antibody disorders, extrapontine osmotic demyelination syndrome, and multiple sclerosis. These diagnoses were ruled out in our patient through appropriate history, physical examination findings, and relevant diagnostic testing.
Two years ago, our patient presented with a 3-week history of intractable nausea, vomiting, and hiccups of unexplained origin, that later subsided without treatment and recurrence. Brain MRI with contrast at the time was unremarkable, although no formal neurological or serological evaluation was pursued.
The area postrema, a circumventricular organ, is an emetogenic center found in the dorsal medulla, at the caudal end of the 4th ventricle [4,12,13]. The area postrema, unlike other areas of the brain affected by AQP-4 IgG antibodies, lacks astrocytic excitatory amino acid transporter 2 (EAAT2), a glutamate transporter channel [12]. Thus, the pathophysiology of area postrema syndrome is more inflammatory than demyelinating, and treatment with steroids and immunotherapy results in a better prognosis [12].
Area postrema syndrome presents as intractable nausea, vomiting, and/or hiccups [1]. Area postrema syndrome is the initial presentation in 12% of NMOSD patients [2]. The criteria for diagnosing area postrema syndrome with AQP-4 seropositivity include acute-subacute episodes of nausea, vomiting, and/or hic-cups with symptoms persisting for a minimum of 48 h, despite symptomatic treatment, and exclusion of other etiologies [14]. In seronegative patients, a dorsal medullary lesion on MRI is required for diagnosis [5,14]. Internists and gastroenterologists should be aware of area postrema syndrome associated with NMOSD to initiate appropriate referrals and prompt treatment [14].
The treatment protocol for acute management for NMOSD includes high-dose steroids (1 g i.v. methylprednisolone daily, followed by oral prednisone taper) [2]. The acute treatment of acute diencephalic syndrome and area postrema syndrome follows similar treatment guidelines, with area postrema syndrome patients showing a more robust symptomatic response compared to other core subsets of NMOSD [14]. Plasmapheresis is the preferred immunomodulatory treatment for patients refractory to steroid treatment [2]. There have been recent advancements in the long-term management of NMOSD, with the approval of 3 immunomodulatory agents, including eculizumab (complement factor C5 inhibitor), inebilizumab (CD-19 inhibitor), and satralizumab (IL-6 receptor inhibitor) [15,16]. Rituximab has also shown a beneficial effect, with a relapse prevention rate of up to 67% [16]. Other commonly used immunosuppressants include azathioprine, mycophenolate mofetil, and tocilizumab [15]. Specific monoclonal antibodies, including ublituximab, which targets CD-20, and aquaporumab, which binds to AQP-4 directly, are in the early stages of development and require large-scale clinical trials before use in clinical practice [15]. In our patient, rituximab was the drug of choice, resulting in no clinical or radiological relapse for 4 months.
Approximately 50% of patients with untreated NMOSD are left with chronic disabilities such as wheelchair dependency [2]. The mortality rate is 33% within 5 years of a NMOSD attack in untreated patients [2]. In our patient, earlier diagnosis at the onset of area postrema syndrome and initiation of appropriate treatment could have prevented the development of acute diencephalic syndrome.
Conclusions
We presented a case of NMOSD in which lack of serological work-up on the initial presentation of intractable nausea, vomiting, and hiccups resulted in delayed diagnosis. Secondly, a broad differential needs to be considered due to a wide range of clinical entities associated with acute diencephalic syndrome. This case report has highlighted the need for increased awareness of NMOSD to be considered as a diagnostic possibility to prevent a potentially debilitating prognosis.
References:
1.. Wingerchuk DM, Banwell B, Bennett JL, International Panel for NMO Diagnosis. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders: Neurology Jul 14, 2015; 85(2); 177-89
2.. Huda S, Whittam D, Bhojak M, Neuromyelitis optica spectrum disorders: Clin Med (Lond), 2019; 19(2); 169-76
3.. Hamid SH, Elsone L, Mutch K, The impact of 2015 neuromyelitis optica spectrum disorders criteria on diagnostic rates: Mult Scler, 2017; 23(2); 228-33
4.. Dutra BG, da Rocha AJ, Nunes RH, Maia ACM, Neuromyelitis optica spectrum disorders: Spectrum of MR imaging findings and their differential diagnosis. [published erratum appears in Radiographics. 2018;38(2):662]: Radiographics, 2018; 38(1); 169-93
5.. Bennett JL, Finding NMO: The evolving diagnostic criteria of neuromyelitis optica: J Neuroophthalmol, 2016; 36(3); 238-45
6.. Kim SM, Kim JS, Heo YE, Cortical oscillopsia without nystagmus, an isolated symptom of neuromyelitis optica spectrum disorder with anti-aquaporin 4 antibody: Mult Scler, 2012; 18(2); 244-47
7.. Kume K, Deguchi K, Ikeda K, Neuromyelitis optica spectrum disorder presenting with repeated hypersomnia due to involvement of the hypothalamus and hypothalamus-amygdala linkage: Mult Scler, 2015; 21(7); 960-62
8.. Khan F, Sharma N, Ud Din M, Bansal V, Isolated Pulvinar/Hockey stick sign in nonalcoholic Wernicke’s encephalopathy: Am J Case Rep, 2020; 21; e928272
9.. Hsu CL, Yeh JH, Lau CI, Persistent hyperthermia in a patient with aquaporin-4-antibody-positive neuromyelitis optica spectrum disorder: J Clin Neurol, 2016; 12(4); 515-16
10.. Jin S, Long Z, Wang W, Jiang B, Hyponatremia in neuromyelitis optica spectrum disorders: Literature review: Acta Neurol Scand, 2018; 138(1); 4-11
11.. Caine D, Halliday GM, Kril JJ, Harper CG, Operational criteria for the classification of chronic alcoholics: Identification of Wernicke’s encephalopathy: J Neurol Neurosurg Psychiatry, 1997; 62(1); 51-60
12.. Iorio R, Lucchinetti CF, Lennon VA, Intractable nausea and vomiting from autoantibodies against a brain water channel: Clin Gastroenterol Hepatol, 2013; 11(3); 240-45
13.. Mirza M, Das J, Neuroanatomy, Area postrema. [Updated 2021 Aug 11]: StatPearls [Internet], 2021, Treasure Island (FL), StatPearls Publishing Available from: https://www.ncbi.nlm.nih.gov/books/NBK544249/
14.. Shosha E, Dubey D, Palace J, Area postrema syndrome: Frequency, criteria, and severity in AQP4-IgG-positive NMOSD: Neurology, 2018; 91(17); e1642-51
15.. Xie Q, Sun M, Sun J, New progress in the treatment of neuromyelitis optica spectrum disorder with monoclonal antibodies (review): Exp Ther Med, 2021; 21(2); 148
16.. Gospe SM, Chen JJ, Bhatti MT, Neuromyelitis optica spectrum disorder and myelin oligodendrocyte glycoprotein associated disorder-optic neuritis: A comprehensive review of diagnosis and treatment: Eye (Lond), 2021; 35(3); 753-68
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Most Viewed Current Articles
07 Dec 2021 : Case report
22,759,422
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  175,936
175,936
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,499
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,510
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133