01 April 2022: Articles 
A Case of Hemophagocytic Lymphohistiocytosis (HLH) Secondary to T Cell Lymphoma and Cytomegalovirus (CMV) Infection and Complicated by Tumor Lysis Syndrome (TLS)
Challenging differential diagnosis, Unusual or unexpected effect of treatment, Diagnostic / therapeutic accidents, Rare disease, Adverse events of drug therapy, Educational Purpose (only if useful for a systematic review or synthesis)
Yassine Kilani1ABDEFG*, Trisha Laxamana1BEG, Kamran Mahfooz1BDEF, Mubarak H. Yusuf1BEG, Victor Perez-Gutierrez1ABEG, Nehad Shabarek1EGDOI: 10.12659/AJCR.935915
Am J Case Rep 2022; 23:e935915






Abstract
BACKGROUND: Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening syndrome of systemic hyper-inflammation, caused by an excessive cytokine secretion, secondary to an uncontrolled proliferation of lymphocytes and macrophages, and leading to vascular endothelial injury and multi-organ failure. HLH is either primary/familial due to genetic mutations in the genes coding for the CD8+ and NK T cells cytotoxic proteins or is secondary to infection, malignancy, or autoimmune disorders. Timely diagnosis using the HLH-2004 criteria and prompt initiation of treatment for HLH is essential for the survival of affected patients. Adults with HLH have poor outcomes even with aggressive treatment.
CASE REPORT: Our patient was a 48-year-old man who presented with altered mental status. He was tachycardic and tachypneic, and quickly developed acute hypoxemic respiratory failure requiring mechanical ventilation. Computed tomography (CT) of the chest and abdomen showed bilateral pleural effusion, ascites, and heterogeneous splenomegaly. Laboratory workup revealed anemia, thrombocytopenia, severe hyperferritinemia, hypofibrinogenemia, and hypertriglyceridemia. Pleural fluid analysis showed a lymphocytic exudate, with T cell predominance on flow cytometry. A T cell rearrangement study of the pleural fluid was positive. Bone marrow biopsy showed histiocytes with hemophagocytic activity. The diagnosis of HLH secondary to T cell lymphoma was made, and the patient was treated with dexamethasone and etoposide. A few hours later, the patient had a cardiac arrest, and laboratory findings suggestive of tumor lysis syndrome (TLS) were discovered. The patient died of refractory shock one day later, and the cytomegalovirus (CMV) PCR result was positive during that day.
CONCLUSIONS: Adults with HLH have poor outcomes even with aggressive treatment. Additional focus on the management of HLH should shift towards preventing complications such as TLS. More studies should focus on post-treatment outcomes of HLH secondary to malignancy to improve the management and prognosis.
Keywords: Cytomegalovirus Infections, Lymphohistiocytosis, Hemophagocytic, Tumor Lysis Syndrome, Adult, Cytomegalovirus, Humans, Lymphoma, T-Cell, Male
Background
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening syndrome of systemic hyper-inflammation. It is caused by an excessive cytokine secretion, secondary to uncontrolled proliferation of lymphocytes and macrophages. The consequences are widespread activation of macrophages, hemophagocytosis, vascular endothelial injury, and subsequent multi-organ failure. HLH is either primary/familial due to genetic mutations or secondary to infections, malignancy, or autoimmune disorders [1,2]. Timely diagnosis using the HLH-2004 criteria and prompt initiation of treatment for HLH are essential for the survival of affected patients. Adults with HLH have poor outcomes even with aggressive treatment [4,5]. Tumor lysis syndrome (TLS) is a well-documented complication of hematologic malignancies treated with cytotoxic chemotherapy. The diagnosis is based on the TLS revised clinical and laboratory criteria [3]. Prevention of TLS rather than treatment improves prognosis.
Case Report
Our patient was a 48-year-old man admitted with altered mental status. He was found by the emergency medical services covered in urines and feces. His roommate stated that he found him confused for the past day, and that he had been bed ridden during the last month with cough and diarrhea. His past medical history was unreliable due to the patient’s confusion.
On admission, his body temperature was 35.8°C and he was tachycardic (118 beats/minute) and tachypneic (28 breaths/minute), with minimal accessory muscle use, and oxygen saturation was 100%. Breath sounds were diminished on the lung bases. The abdomen was soft, distended, but non-tender. Bilateral pitting edema was present in the feet. The rest of the examination was normal. Hours after his admission in the emergency room, he developed acute hypoxemic respiratory failure with tachypnea up to 40 breaths/min, and an oxygen saturation 85% on room air. He was intubated and transferred to intensive care.
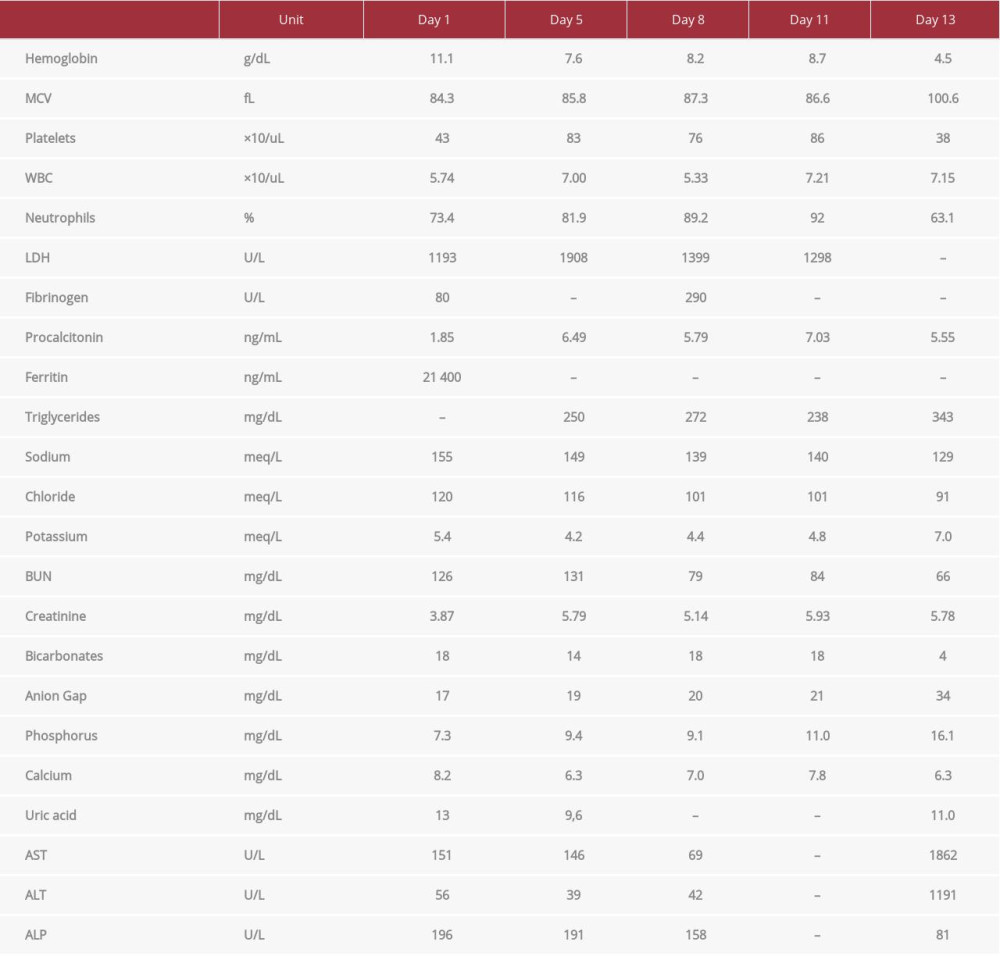
Computed tomography (CT) of the chest and abdomen revealed bilateral pleural effusions, a small pericardial effusion, a large heterogeneous splenomegaly, ascites, and mesenteric and soft-tissue anasarca (Figure 1). A head CT showed no intracranial pathology, with diffuse cerebral atrophy (out of proportion to the patient’s age). A summary of the laboratory findings is provided in Table 1. The severe hyperferritinemia prompted us to consider differential diagnoses such as iron overload, hepato-cellular disease, kidney disease, malignancy, sepsis, infections (viral, fungal), and inflammatory/rheumatologic conditions. A limitation of this case report is the lack of diagnostic testing for iron overload (transferrin saturation). However, given the acuity of the presentation, our differential was narrowed down to sepsis, infectious etiologies, and HLH. Blood, sputum, and urine cultures were negative. Hepatitis B surface antigen (HBsAg) and antibody (HBsAb), hepatitis C antibody, Epstein-Barr virus (EBV) PCR, SARS-CoV-2 RNA PCR, HIV 1,2 antigen/antibody test, and HTLV-1/2 antibodies were non-reactive. PCR for cytomegalovirus (CMV) DNA was ordered. Autoimmune workup with antinuclear antibodies, anti-double stranded DNA, Beta-2 Glycoprotein 1 antibody, and C3 and C4 complement was negative. Diagnostic and therapeutic paracentesis and thoracentesis were performed on day 2. Pleural fluid was exudative (pleural LDH 549 U/L) and lymphocytic (3600 white blood cells and 86% lymphocytes). Cytology of the pleural and ascitic fluids showed macrophages and inflammation and was negative for malignant cells. Flow cytometry of the pleural fluid showed T cell predominance (positive CD3, CD5, CD4, partial CD8, negative CD10, rare to absent positive B cells CD19/CD20), and did not detect T/B cell lymphoma. A T cell rearrangement study of the pleural fluid was positive, raising the suspicion for T cell lymphoma. A repeat chest CT was suggestive of bilateral pneumonia. Bone marrow biopsy revealed histiocytes with hemophagocytic activity (Figure 2), without evidence of leukemia, lymphoma, plasma cell carcinoma, or metastatic carcinoma. Additional testing with Interleukin 2 Receptor CD25 Soluble was elevated to 27 416 pg/mL, confirming the diagnosis of HLH as per the HLH-2004 criteria. The diagnosis of HLH secondary to T cell lymphoma was the most likely.
Our patient was treated with fluids, vasopressors, and broad-spectrum antibiotics for suspicion of aspiration pneumonia. Worsening renal function and anasarca prompted the initiation of renal replacement therapy on day 5. The HLH-94 protocol with dexamethasone 20 mg daily and renally adjusted etoposide 75 mg/m2 was initiated on day 12. Ten hours later, the patient had a cardiac arrest secondary to pulseless electrical activity, with return of spontaneous circulation (ROSC) after 7 min. One day later, he developed refractory shock and died. Laboratory workup after ROSC revealed severe hyperkalemia 7.0 meq/L, hyperphosphatemia 16.1 mg/dL, hypocalcemia 6.3 mg/dL, hyperuricemia 11.0 mg/dL, and elevated blood urea nitrogen 66 mg/dL and creatinine 5.78 mg/dL. We presume that the cardiac arrest was likely secondary to the severe electrolytes abnormalities (Table 1). The sudden worsening of laboratory abnormalities hours after chemotherapy raised the suspicion of TLS. CMV PCR test results came back positive at 6900 IU/mL (N <96 IU/mL) on the same day.
Discussion
We report a case of HLH likely secondary to T cell lymphoma and CMV infection, treated with dexamethasone and etoposide, and complicated by TLS. HLH is characterized by systemic hyper-inflammation, secondary to an uncontrolled proliferation of natural killer (NK) cells, CD8+ T cells, and macrophages, causing cytokine hyper-secretion, widespread activation of macrophages, hemophagocytosis, endothelial injury, and multi-organ failure. HLH can be either primary or secondary. Primary HLH is due to familial genetic mutations in the genes coding for the CD8+ and NK T cells cytotoxic proteins (PRF1, STX11, STXBP2, and UNC13D) [2]. Secondary HLH occurs after infections, malignancy, and autoimmune disorders or is idiopathic [5–7].
HLH is a rare disease and the true incidence is currently difficult to determine in the literature. However, since the institution of the HLH-2004 diagnostic criteria, the disease is now more recognized. While primary HLH presents early in childhood, secondary HLH is more common in adults. The annual incidence of primary HLH was 1.2–1.5 per million children per year according to the Swedish National Registry [8]. The mean age of adult HLH was found to be 49 years, with a male predominance (sex ratio of 1.9) [9]. Our patient had evidence of T cell lymphoma. In the literature, 1% of patients with hematologic malignancies were diagnosed with HLH [10]. We suspected HLH based on the extreme elevation of ferritin. The differential diagnosis for severe hyperferritinemia includes iron overload, hepatocellular or kidney disease, malignancy, sepsis, and inflammatory conditions (HLH, macrophage activation syndrome [MAS], Still’s disease, and other autoimmune conditions) [11,12]. However our patient’s acute presentation was more suggestive of sepsis, infection, or acute inflammation. The diagnosis of HLH is either molecular (mutations consistent with familial or primary HLH) or made by the presence of at least 5 of the following 8 criteria: splenomegaly, fever, cytopenias in ≥2 of 3 lineages (hemoglobin <9.0 g/dL, platelets <100 000/uL, neutrophils <1000/uL), ferritin >500 ng/mL, triglycerides >265 mg/dL, and/or fibrinogen <150 mg/dL, hemophagocytosis on the bone marrow or spleen or lymph node, low or absent NK cell activity, and soluble IL-2 receptor elevated (sCD25) ≥2400 U/mL [4]. Our case was diagnosed based on 6 of the 8 HLH-2004 criteria.
The current standard of care, the HLH-94 protocol, consists of an 8-week initial therapy with immunosuppressive and cytotoxic therapy, followed by continuation therapy, for a total of 52 weeks. Initial therapy includes dexamethasone and etoposide (VP-16), with cyclosporine A (CSA) added on week 9. Intrathecal methotrexate (IT-MTX) is indicated on week 3 in case of progressive neurological symptoms or in case of abnormal cerebrospinal fluid value. Hematopoietic stem cell transplantation (HSCT) is indicated in patients with familial/genetic, relapsing, or severe/persistent disease, and should be performed as soon as a suitable donor is available [13,14]. The HLH-2004 protocol reduced the total duration of the treatment to 40 weeks and added cyclosporine to the initial therapy, and intrathecal steroids to IT-MTX in case of neurologic dysfunction. However, no statistically significant improvement was noted [13].
Timely recognition of signs of cytokine excess and treatment of HLH in acutely ill patients is essential for patient survival. Indeed, adults with HLH have poor outcomes even with aggressive treatment [5]. While etoposide treats HLH by inhibiting monocyte-macrophage activation [15], it is one of the cytotoxic chemotherapies most associated with TLS, especially in the context of hematologic malignancy [16]. TLS is best prevented rather than managed. Aggressive hydration, removal of nephrotoxic drugs (iodinated contrast, non-steroidal anti-inflammatory drugs), prophylactic xanthine oxidase inhibitors (allopurinol), and urate oxidase inhibitor therapy (rasburicase) have been shown to improve outcomes [3]. Despite improvement in mortality rates, the prognosis remains guarded [17].
Conclusions
Adults with HLH have poor outcomes even with aggressive treatment. Additional focus on the management of HLH should shift towards preventing complications such as TLS. More studies should focus on post-treatment outcomes of HLH secondary to malignancy to improve the management and prognosis.
References:
1.. Janka GE, Lehmberg K, Hemophagocytic syndromes – an update: Blood Rev, 2014; 28(4); 135-42
2.. Zhang K, Astigarraga I, Bryceson Y, Familial hemophagocytic lymphohistiocytosis: GeneReviews® [Internet], 2006; 1993-2022, Seattle (WA), University of Washington Seattle
3.. Howard SC, Jones DP, Pui CH, The tumor lysis syndrome [published correction appears in N Engl J Med. 2018;379(11): 1094]: N Engl J Med, 2011; 364(19); 1844-54
4.. Henter JI, Horne A, Aricó M, HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis: Pediatr Blood Cancer, 2007; 48(2); 124-31
5.. Al-Samkari H, Berliner N, Hemophagocytic lymphohistiocytosis: Annu Rev Pathol, 2018; 13; 27-49
6.. Schram AM, Comstock P, Campo M, Haemophagocytic lymphohistiocytosis in adults: A multicentre case series over 7 years: Br J Haematol, 2016; 172(3); 412-19
7.. Morimoto A, Nakazawa Y, Ishii E, Hemophagocytic lymphohistiocytosis: Pathogenesis, diagnosis, and management: Pediatr Int, 2016; 58(9); 817-25
8.. Meeths M, Horne A, Sabel M, Bryceson YT, Henter JI, Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden: Pediatr Blood Cancer, 2015; 62(2); 346-52
9.. Parikh SA, Kapoor P, Letendre L, Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis: Mayo Clin Proc, 2014; 89(4); 484-92
10.. Machaczka M, Vaktnäs J, Klimkowska M, Hägglund H, Malignancy-associated hemophagocytic lymphohistiocytosis in adults: A retrospective population-based analysis from a single center: Leuk Lymphoma, 2011; 52(4); 613-19
11.. Sandnes M, Ulvik RJ, Vorland M, Reikvam H, Hyperferritinemia – a clinical overview: J Clin Med, 2021; 10(9); 2008
12.. Sackett K, Cunderlik M, Sahni N, Extreme hyperferritinemia: Causes and impact on diagnostic reasoning: Am J Clin Pathol, 2016; 145(5); 646-50
13.. Henter JI, Aricò M, Egeler RM, HLH-94: A treatment protocol for hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte Society: Med Pediatr Oncol, 1997; 28(5); 342-47
14.. La Rosée P, Horne A, Hines M, Recommendations for the management of hemophagocytic lymphohistiocytosis in adults: Blood, 2019; 133(23); 2465-77
15.. Zhou H, Yang M, Cui L, Jiang J, Chimeric antigen receptor T cell therapy and nephrotoxicity: From diagnosis to treatment strategies: Int Immunopharmacol, 2020; 89(Pt B); 107072
16.. Tosi P, Barosi G, Lazzaro C, Consensus conference on the management of tumor lysis syndrome: Haematologica, 2008; 93(12); 1877-85
17.. McBride A, Trifilio S, Baxter N, Managing tumor lysis syndrome in the era of novel cancer therapies: J Adv Pract Oncol, 2017; 8(7); 705-20
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Most Viewed Current Articles
07 Dec 2021 : Case report
22,759,422
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  175,936
175,936
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,499
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,510
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133