03 August 2022: Articles 
Management of Patients with Alagille Syndrome Undergoing Living Donor Liver Transplantation: A Report of 2 Cases
Rare disease
Odai Jamaan Alqahtani1ABCDEFG*, Riaz Nazeer Ahmad1ADFG, Abdullah Bakr Abolkhair1ADEFG, Aljazi Dhari Alrashid1ABCDEFGDOI: 10.12659/AJCR.936513
Am J Case Rep 2022; 23:e936513






Abstract
BACKGROUND: Alagille syndrome (AGS) is a rare genetic disease characterized by 5 typical features: peculiar facial anomaly, posterior embryotoxon, chronic cholestasis, butterfly-like vertebral-arch defects, and cardiovascular malformations. AGS in a liver transplant setting is particularly rare in Saudi Arabia. This case report presents successful anesthetic management in AGS patients during liver transplantation surgery.
CASE REPORT: We present here 2 patients with AGS who underwent liver transplantation surgery. Case 1 describes a 3-year-old boy who was diagnosed with AGS at the age of 2 weeks, manifesting as a prominent forehead, deep-set eyes, pointed chin, and butterfly-shaped vertebrae, along with coarctation of the aorta, peripheral branch pulmonary artery stenosis, direct hyperbilirubinemia, cholestasis, and G6PD deficiency. Case 2 describes a 12-year-old girl, known to have AGS, who presented with decompensated liver disease, portal hypertension, splenomegaly, hypersplenism, and portal vein thrombosis, as well as the other dysmorphic features of AGS, such as a prominent forehead, deep-set eyes, pointed chin, and butterfly-shaped vertebrae. The anesthetic management of the 2 patients started from the perioperative period with careful assessment and optimization of the cardiovascular system, intraoperative maintenance of stable hemodynamics, the use of inhaled nitric oxide when clinically indicated (used in case 1), and the use of goal-directed management of fluid as well as blood and blood products. These all played a major role in the successful management of anesthesia for these patients.
CONCLUSIONS: The important features of successful anesthetic management included thorough preoperative cardiovascular system evaluation and intra-operative maintenance of normal temperature, ionized calcium, pH, hemoglobin, and stable hemodynamics.
Keywords: Alagille Syndrome, Liver Transplantation, Anesthetics, Child, Child, Preschool, Cholestasis, Female, Humans, Infant, Newborn, Living Donors, Male
Background
Alagille syndrome (AGS) is a rare genetic disease, with an autosomal dominant inheritance pattern, that results from mutations in the JAG1 or NOTCH2 genes [1]. AGS is a multisystem disorder characterized by bile duct anomaly, congenital cardiac defects, butterfly vertebrae, posterior embryotoxon of the eye, and typical facial features. Other associated features include growth retardation, neurovascular events, and kidney disease. The hepatic involvement is characterized by insufficiency of intrahepatic bile ducts resulting in hyperbilirubinemia, intrahepatic cholestasis, and hypercholesterolemia [2]. The disease has an estimated prevalence rate of 1 in 30,000 to 1 in 70,000 live births [1,3].
Evidently, the management of a multisystem disease like AGS warrants a multidisciplinary approach and may require the involvement of a gastroenterologist, cardiologist, ophthalmolo-gist, nephrologist, hepatologist, liver transplant specialist, geneticist, nutritionist, and child development expert, depending on the age and specific organ defects of the patient [3]. The hepatic involvement presents a particularly challenging situation as no form of phenotypic or genotypic features can predict the development of end-stage liver disease. Liver transplant remains the last resort for many of these AGS patients with end-stage liver disease [4].
Case Reports
CASE 1:
A 3-year-old boy had been diagnosed at the age of 2 weeks with AGS, which manifested as a prominent forehead, deep-set eyes, pointed chin, and butterfly-shaped vertebrae along with coarctation of the aorta, peripheral branch pulmonary artery stenosis, direct hyperbilirubinemia, cholestasis, and G6PD deficiency. The baby had been delivered by Cesarean section, with a birth weight of 2.2 kg. His mother’s obstetric history was G3P3. In the postnatal period, the patient developed neonatal jaundice, for which he was kept in the nursery for 3 days.
His mother was a known carrier of sickle cell trait, G6PD, and alpha thalassemia, and his father was a known carrier of sickle cell trait. His grandparents were also carriers of sickle cell trait. His 2 siblings were normal, with no cardiac anomaly detected. On genetic testing, he was heterozygous for a JAK1 gene mutation and homozygous for a G6PD mutation. After worsening of cardiac symptoms and cholestasis, the patient was transferred to a specialist center (King Faisal Specialist Hospital And Research Center, Riyadh) for further management and cardiac surgery.
A preprocedural echocardiogram was performed and showed mild narrowing of the aortic isthmus with a peak gradient of 16 mmHg, without diastolic drag. The other findings noted were: small right pulmonary artery (RPA) with a peak gradient of 36 mmHg with diastolic drag, and normal-sized left pulmonary artery (LPA) with turbulent flow, peak gradient of 22 mmHg with normal-sized cardiac chambers and biventricular systolic function. Abdominal aortic Doppler flow was normal, and there was no right or left ventricular outflow tract obstruction.
For the pulmonary artery stenosis, the patient underwent an RPA dilatation procedure. Post-dilatation of RPA, right ventricular systemic blood pressure still remained at 60% of the pre-dilatation pressure. The pullback of the catheter from the RPA to the main pulmonary artery showed no systolic gradient. Angiograms showed significantly improved RPA diameter and no dissection or aneurysm was noticed. Post-dilatation, the proximal RPA measured 7.36 mm in diameter.
A post-procedure echocardiogram was performed which showed normal-sized RPA with no stenosis and an LPA with mildly increased velocity and a peak gradient of 30 mmHg. Biventricular systolic function was normal with no pericardial effusion.
One year after the echocardiogram procedure, a small RPA was visible, with a distal stenosis peak gradient of 23 mmHg and distal gradient (intrapulmonary pressure of 31 mmHg). A normal-sized proximal RPA and mild distal LPA stenosis with peak gradients of 22 mmHg were noted. The proximal LPA was normal-sized but the right ventricle was hypertrophied and mildly dilated. A right ventricular pressure of 46% with systemic post-RPA dilation was noted. There was no right or left ventricular outflow tract obstruction and the biventricular systolic function was normal. Mild turbulence was seen in the descending thoracic aorta but no real coarctation. The abdominal aortic Doppler flow was normal.
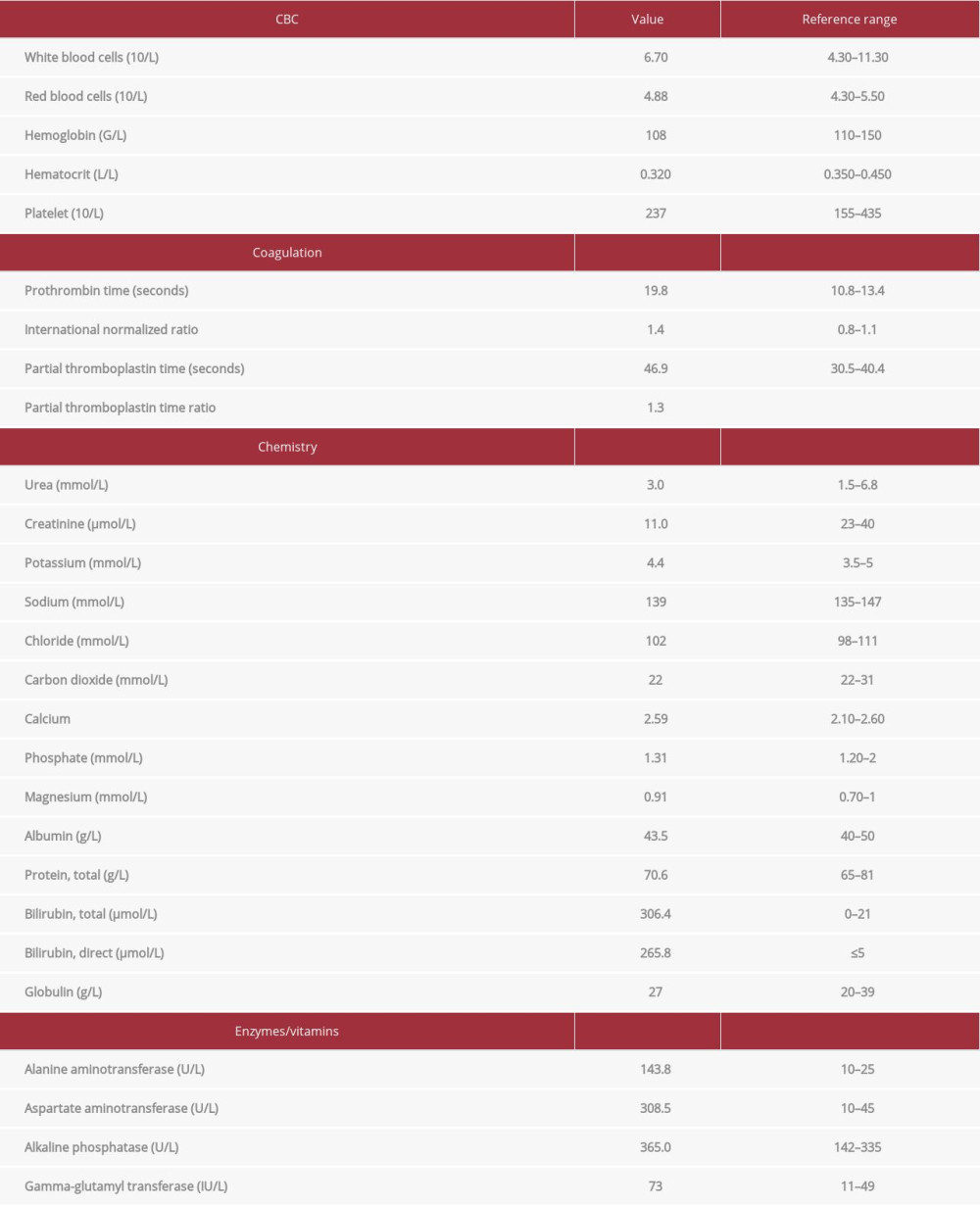
After reaching this stage of cardiac optimization and considering the patient’s liver disease secondary to AGS, which had a calculated model for end-stage liver disease (MELD) score of 21, the cardiology team agreed on liver transplant surgery as the course of action. The liver donor for this patient was the patient’s paternal uncle. His weight and height at the time of surgery were 13.7 kg and 96 cm respectively, and his preoperative laboratory results as well as preoperative liver ultra-sound are shown in Table 1 and Figure 1.
In the operating room, anesthesia was induced through intravenous (IV) administration, using midazolam 1 mg, propofol 40 mg, fentanyl 30 mcg, and atracurium 10 mg. Intubation with a direct laryngoscope using a regular size-5 cuffed endotracheal tube (ETT) was done. Sevoflurane was used for maintenance of anesthesia as well as a continuous IV infusion of fentanyl 27.4 mcg/h and atracurium 6.8 mg/h.
For hemodynamic stability during the operation, a right radial arterial line, a right femoral arterial line, and a left cephalic triple lumen central line were placed. Hemodynamics were maintained using a milrinone 0.05 mcg/kg/min infusion and norepinephrine at a minimum infusion rate. Nitric oxide at 20 ppm was used to reduce pulmonary vascular resistance and was stopped before reperfusion. The surgery performed was a living related donor liver transplant using a left lateral segment to generate a 285-gram graft. During surgery, 115 ml of packed red blood cells (PRBCs) were given, in addition to 500 ml of normal saline and 300 ml of 5% albumin. Total blood loss was around 300 ml, and urine output was 400 ml.
After the surgery, all anesthetic agents were switched off, neostigmine and glycopyrrolate were given to reverse the muscle relaxant, and the patient was extubated and transferred to the pediatric intensive care unit (PICU) with no anesthesia complications, in stable condition, and without the need for vasoactive medications. The total time for the surgery was around 8 hours. The patient remained in the PICU for postoperative care for around 1 week with continuous monitoring and close observation of the liver function without any significant complications. After the week in the PICU, he was transferred to the ward floor for another week. Then, he was discharged from the hospital with regular followup in the hepatic transplant clinic.
CASE 2:
A 12-year-old girl known to have AGS presented with decompensated liver disease, portal hypertension, splenomegaly, hypersplenism, and portal vein thrombosis. The other dysmorphic features of AGS such as prominent forehead, deep-set eyes, pointed chin and butterfly-shaped vertebrae were also present in this case.
She was diagnosed with AGS at the age of 6 and was kept on ursodiol and vitamins. The patient had 4 siblings. All were healthy and alive. She had no family history of any liver disease. During this period, she was on oral alfacalcidol, oral lactulose, multivitamins, phytonadione, ursodiol, and vitamin E. For the above complaints, liver transplantation was planned. At the age of 12, the patient’s mother was informed by the primary physician that the patient’s liver was decompensated and she must be referred to a specialist center (King Faisal Specialist Hospital And Research Center, Riyadh) for evaluation of liver disease and management.
Preoperative cardiology assessment showed a normal cardiac shadow on chest x-ray, normal sinus rhythm on electrocardiogram (ECG), small and stenotic LPA, measuring 5.2 mm, Z score of 3.8, and estimated LPA peak gradient of 25 mmHg with normal-sized RPA. A small patent foramen ovale with a left to right shunt was noted, with an intact ventricular septum. There was no patent ductus arteriosus.
A competent mitral valve with a mean/peak gradient of 3/7 mmHg was seen. There was mild tricuspid valve regurgitation, but no stenosis of the tricuspid valve. No left or right ventricular outflow tract obstructions were seen.
The chambers were normal-sized with normal biventricular systolic function and systemic and pulmonary venous drainage, and no coarctation on echocardiogram.
The opinion of the cardiology team, taken preoperatively, was that there was no contraindication for liver transplantation as the right ventricular pressure was low and only unilateral LPA stenosis was seen. A V/Q scan was recommended, to assess perfusion to the left lung and followup with the pediatric cardiology clinic was recommended, as the LPA might need intervention later.
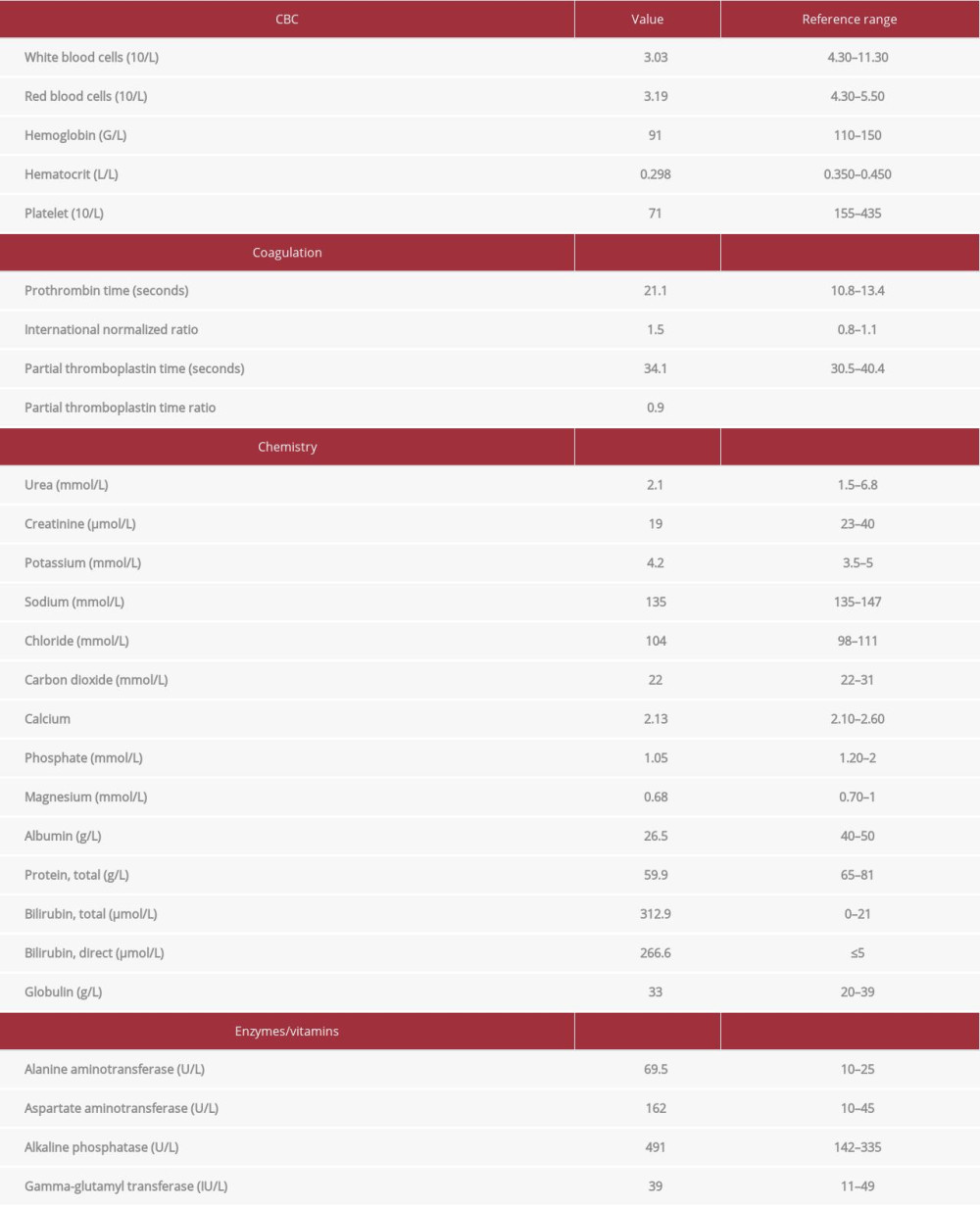
The decompensated liver and cholestasis, due to AGS, had a calculated MELD score of 23. The patient underwent liver transplantation and the donor in this case was the mother of the patient. Her weight and height at the time of surgery were 24.4 kg and 129 cm, respectively, and her preoperative laboratory results as well as preoperative liver ultrasound are shown in Table 2, Figures 2, 3.
Intraoperative anesthesia was induced through IV administration using midazolam 2 mg, propofol 80 mg, fentanyl 75 mcg, and atracurium 20 mg, and intubation was carried out using a direct laryngoscope with a size 5 cuffed ETT. The anesthesia was maintained using sevoflurane as well as IV infusion of fentanyl (48 mcg/h) and atracurium (12.2 mg/h).
To maintain hemodynamic stability during the operation, the left femoral arterial line and right radial arterial line, right cephalic triple lumen central line, 2 peripheral intravenous catheters (18G), and a nasogastric tube were placed. Hemodynamic maintenance was carried out using a norepinephrine infusion at a minimum infusion rate of 0.05 mcg/kg/min. The left lateral lobe, with a weight of 299 g, was used for the liver transplant.
After the operation, the patient was shifted to the PICU in stable condition and extubated without inotropic support. There was no significant bleeding, with an estimated blood loss of 200 ml. One unit of PRBC was transfused to compensate for this blood loss. Urine output was 500 ml. Crystalloids (500 ml) were given, and the total time of the surgery was around 6 hours. In the PICU, the patient was monitored with close observation of her liver function. She did not require inotropic support and there were no major complications during her stay in the PICU. She was shifted to the normal ward within 1 week and discharged from the hospital 2 weeks after the surgery.
Postoperative echocardiography conducted 3 weeks after the surgery showed sinus tachycardia and a small, closing, patent foramen ovale with a minimal left to right shunt. A small LPA, measuring a peak gradient of 14 mmHg with normalsized RPA, was noted. The mitral valve was competent, with a peak/mean gradient of 8/3 mmHg. The chambers were normal-sized with normal biventricular systolic function. No coarctation was noted.
Discussion
In 1969, Dr. Daniel Alagille, a French pediatric gastroenterologist, was the first to describe a syndrome comprising typical facies, intrahepatic bile duct insufficiency and cholestasis, heart murmur, butterfly vertebrae, and posterior embryotoxon [5]. AGS was initially defined by Watson [6] and Allagille [7]. This multisystem disease is characterized by 5 clinical features: chronic cholestasis, typical facial anomaly, posterior embryo-toxon of eye, vertebral-arch defects, and cardiovascular malformations [6,7].
Multisystem involvement can have short- or long-term prognostic significance and hence, thorough consideration is required during pre-liver transplantation evaluation [8]. The intensity of systemic involvement might be variable even within families, which can lead to diagnostic errors [7].
AGS patients with severe hepatic manifestations have a poor quality of life because refractory pruritus invariably leads to disturbances in sleep and failure in growth and thus consequences of impairment in school and social achievement [9]. Around 25% of AGS patients may need liver transplantation in childhood [3,8,10]. Most of the AGS patients that require liver transplantation have complications of cholestasis, severe hypercholesterolemia, and osteodystrophy. Previous literature has shown 1-year survival after liver transplantation to range from 71% to 100%. The severity of cardiac anomalies is the deciding factor for the success rate of liver transplantation. Pretransplantation cardiac evaluation must be done properly. In addition to intra-operative difficulties, the long-term usage of antirejection therapy drugs can cause atherosclerosis, nephrotoxicity, and bone changes. Some studies have reported, however, that pulmonary stenosis and right ventricular hypertrophy did not lead to any intraoperative problems during liver transplantation [11].
AGS is considered syndromic in the presence of all 5 features. Four or fewer features are seen in incomplete or partial forms [12]. Medical treatment comprises nutritional supplements of medium-chain triglycerides, fat-soluble vitamins, and essential fatty acids. There are many published reports of successful liver transplantation in patients with liver failure or portal hypertension [9]. Cardiac defects have been observed in approximately 85% to 100% of AGS cases [10]. The most frequent anomaly is a nonprogressive form of pulmonary stenosis, with varying severity [6]. The mortality rate in AGS as a result of cardiopulmonary complications is around 10%. Greater mortality rates after liver transplantation are observed in AGS patients in comparison with non-AGS patients. Hence, it becomes crucial to do a careful and thorough preoperative cardiac evaluation. In cases of severe cardiac defects that can be treated with cardiac surgery or balloon angioplasty, treatment of the defects should be performed before liver transplantation [12].
Both of our cases suffered from a syndromic form of AGS consisting of cholestasis linked with peripheral pulmonary artery stenosis and peculiar facial features. One of our patients also had a mild form of aortic stenosis. Due to severe jaundice with pruritus, retardation in growth, and pulmonary stenosis, liver transplantation was required. One case needed preoperative treatment; specifically, correction for pulmonary artery stenosis.
Two-dimensional echocardiography is the main method of investigation for early diagnosis and followup of congenital heart disease, with improved 3-dimensional echocardiography as well, when available [13]. As congenital heart disease is highly prevalent, any child with suspected AGS must be fully evaluated with a cardiac check-up, complete history, and physical examination by a pediatric cardiologist. A 12-lead electrocardiogram and a transthoracic echocardiogram are used for assessment of repolarization abnormalities arising due to hepatic dysfunction. Regardless of AGS diagnosis, the recommended evaluations are crucial for any form of liver disease for which liver transplantation is being considered [14]. In both of the present cases, pre- and post-operative echocardiography was conducted to evaluate congenital cardiac defects associated with AGS.
It is recommended to avoid banked blood products and to instead infuse crystalloids or commercial colloid solutions for maintaining adequate intravascular volume to reduce acute blood loss. Central venous pressure of about 10 cmH2O under stable hemodynamic conditions can be sustained in anemic cases with the administration of 5% albumin and 11.2 ml/kg/h crystalloids. Ionized calcium and arterial blood gases must be evaluated on a routine basis during liver transplantation for the prevention of citrate toxicity, which can lead to ionized hypocalcemia. Citrate toxicity can result from increased exogenous citrate load after transfusion of blood products during liver transplantation. Our patient needed calcium chloride administration because of the use of 5% albumin. The albumin becomes bound to ionized calcium and can lead to hypo-ionized calcemia and fluctuating hemodynamics [12].
The operating room temperature was 24°C instead of 19–21°C. This allowed us to sustain the body temperature within the normal range. Normothermia during liver transplantation is a very important factor to prevent complications like coagulopathy and thromboembolic events. Coagulopathies usually occur in patients who experience hypothermia, even with normal values of clotting factors, prothrombin time, and activated partial thromboplastin time, as the low temperature can directly block the enzymatic coagulation cascade reactions. Hypothermia can reduce the activity of platelets due to lower thromboxane B production, which exaggerates cold-induced thrombocytopenia, bone marrow suppression, and hepatosplenic sequestration. Hypothermia commonly leads to higher transfusion requirements and blood loss, and is seen mainly in major operations like liver transplantation [10,12]. In our patient, we maintained normothermia throughout the operation by warming the transfused fluids and using blankets.
As AGS patients have multisystem involvement, they must undergo complete evaluation before liver transplantation. Continuous cholestasis, bone fractures, severe xanthomatosis, refractory pruritus, and growth retardation are the main liver transplantation indications [5,16]. In our patients, liver transplantation was indicated due to jaundice with pruritus and growth retardation. The success rate of liver transplantation is dependent on careful preoperative and perioperative multi-disciplinary planning and management. The pediatric cardiology team suggested that no preoperative correction or treatment was required for one case before liver transplantation. Intraoperative central venous pressure of about 10 mmHg and stable hemodynamics were sustained with adequate fluid resuscitation and vasopressor infusion [10]. Both patients were discharged from the hospital after 2 weeks but the long-term prognosis is unpredictable because of the effects of immunosuppressive treatment on vascular and kidney disease.
A particularly fatal complication after liver transplantation is hepatic artery thrombosis. This complication causes high morbidity and mortality in pediatric patients with a small hepatic artery [8]. Polycythemia cases should be avoided as this is a cause of hepatic artery thrombosis. It is important to maintain a hemoglobin value of approximately 7–9 g/dl so as to create the rheological effects of hemodilution and thus avoid hepatic artery thrombosis.
Conclusions
To conclude, we hereby present the successful anesthetic management of 2 AGS patients with low hemoglobin levels who underwent living liver transplantation with blood product transfusion. The important features of successful anesthetic management included thorough preoperative cardiovascular system evaluation and intraoperative maintenance of normal temperature, ionized calcium, pH, and stable hemodynamics. The hemoglobin level was maintained through blood transfusion. The surgical blood loss was adequately replaced by 300 ml 5% albumin and crystalloids. Anemia was well-tolerated by our patients and it did not delay or affect postoperative recovery or success of the new liver allograft.
Figures
References:
1.. Valamparampil JJ, Reddy MS, Shanmugam N, Living donor liver transplantation in Alagille syndrome – single center experience from south Asia: Pediatr Transplant, 2019; 23(8); e13579
2.. Flores CD, Yangyang RY, Miloh TA, Surgical outcomes in alagille syndrome and PFIC: A single institution’s 20-year experience: J Pediatr Surg, 2018; 53(5); 976-79
3.. Kamath BM, Schwarz KB, Hadžić N, Alagille syndrome and liver transplantation: J Pediatr Gastroenterol Nutr, 2010; 50(1); 11-15
4.. P Singh S, K Pati G, Alagille syndrome and the liver: Current insights: Euroasian J Hepatogastroenterol, 2018; 8(2); 140-47
5.. Alagille D, Habib EC, Thomassin N, L’atresie des voies biliaires intrahepatiques avec voies biliaires extrahepatiques permeables chez l’enfant: J Par Pediatr, 1969; 301; 301-18
6.. Watson GH, Miller V, Arteriohepatic dysplasia: Familial pulmonary arterial stenosis with neonatal liver disease: Arch Dis Child, 1973; 48(6); 459-66
7.. Alagille D, Odievre M, Gautier M, Dommergues JP, Hepatic ductular hypoplasia is associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur: J Pediatr, 1975; 86; 63-71
8.. Kamath BM, Yin W, Miller H, Outcomes of liver transplantation for patients with Alagille syndrome: The studies of pediatric liver transplantation experience: Liver Transpl, 2012; 18(8); 940-48
9.. Cardona J, Houssin D, Gauthier F, Liver transplantation in children with Alagille syndrome – a study of twelve cases: Transplantation, 1995; 60; 339-42
10.. Karaaslan P, Özgür S, Yanaral TU, Living-related donor liver transplantation in a patient with alagille’s syndrome with severe pulmonary stenosis: J Gastroenterol, 2016; 1(2); 3
11.. Ozçay F, Varan B, Tokel K, Severe peripheral pulmonary stenosis is not a contraindication to liver transplantation in Alagille syndrome: Pediatr Transplant, 2006; 10(1); 108-11
12.. Cheng KW, Huang CJ, Wang CH, Anesthetic management of a patient with Alagille’s syndrome undergoing living donor liver transplantation without blood transfusion: Chang Gung Med J, 2004; 27(6); 449-53
13.. Simpson J, Lopez L, Acar P, Three-dimensional echocardiography in congenital heart disease: An expert consensus document from the European Association of Cardiovascular Imaging and the American Society of Echocardiography: J Am Soc Echocardiogr, 2017; 30(1); 1-27
14.. Tretter JT, McElhinney DB, Cardiac, aortic, and pulmonary vascular involvement in Alagille syndrome: Alagille syndrome, 2018; 77-90, Springer, Cham
15.. Tzakis AG, Reyes J, Tepetes K, Liver transplantation for Alagille’s syndrome: Arch Surg, 2012; 128(3); 337-39
16.. Vajro P, Ferrante L, Paolella G, Alagille syndrome: An overview: Clin Res Hepatol Gastroenterol, 2012; 36(3); 275-77
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133