03 November 2024: Articles 
Finerenone as a Novel Treatment for Gitelman Syndrome: A Case Study of a 35-Year-Old Male with Adrenal Mass and Hypokalemia
Challenging differential diagnosis, Rare disease, Adverse events of drug therapy
Rujie Jiang1ABCDEF, Qiue Liu1B, Yanan Sun1B, Xiaoqing Dai1C, Feng Xu1D*, Fang Wang1ADEFDOI: 10.12659/AJCR.944492
Am J Case Rep 2024; 25:e944492






Abstract
BACKGROUND: Gitelman syndrome (GS) is an autosomal recessive salt-losing tubulopathy characterized by renal potassium loss, hypokalemia, metabolic alkalosis, hypocalciuria, hypomagnesemia, and hyper-reninemic hyperaldosteronism. Finerenone is a non-steroidal mineralocorticoid receptor antagonist that inhibits receptor-mediated sodium reabsorption and decreases receptor overactivation. This report describes a 35-year-old man with hypokalemia, a mass in the right adrenal gland, and a diagnosis of Gitelman syndrome with a c.1456>A heterozygous variant of the SLC12A3 gene, treated with finerenone.
CASE REPORT: A 35-year-old man was admitted to the affiliated Hospital of Qingdao University because of a mass in the right adrenal gland. He was in generally good condition upon admission. He was a non-smoker and non-drinker. The examination at admission led to diagnosis of severe hypokalemia. Genetic tests showed that he carried a homozygous pathogenic variant c.1456>A in SLC12A3, which can confirm the diagnosis of Gitelman syndrome. Spironolactone was used to increase the blood potassium level, but after adverse effects were noted, finerenone was used, which greatly improved his blood potassium levels.
CONCLUSIONS: For patients with Gitelman syndrome who cannot tolerate adverse effects such as sex hormone-related adverse reactions from using non-selective mineralocorticoid receptor antagonists, especially male patients, finerenone may be considered as an adjunct therapy for potassium retention and magnesium supplementation. To the best of our knowledge, this is the first report in the world of using finerenone to treat Gitelman syndrome. This provides more options for treatment of patients in the future.
Keywords: finerenone, Gitelman syndrome, Hypokalemia, Mutation, Humans, Male, adult, Mineralocorticoid Receptor Antagonists, Solute Carrier Family 12, Member 3, Naphthyridines
Introduction
Gitelman syndrome (GS) is a rare autosomal recessive, salt-wasting, renal tubular disease, also known as familial hypokalemia and hypomagnesemia, which was first reported by Gitelman in 1966. The main clinical features are hypokalemia, hypomagnesemia, metabolic alkalosis, hypocalciuria, and normal or low blood pressure, and it is more often diagnosed in adolescence or adulthood. GS is usually caused by mutations in the SLC12A3 gene encoding the thiazide diuretic-sensitive sodium-chloride cotransporter (NCC) located in the renal distal tubule [1]. It is usually treated with magnesium, sodium, and calcium. GS is considered one of the most common inherited renal tubular diseases [1]. Patients with this disease often experience transient muscle weakness and tetany, but some are asymptomatic or have relatively mild or nonspecific symptoms, so in most cases they are discovered incidentally during biochemical tests [2]. The prevalence of this disease is about 25 per 1 million population. There are no data on the prevalence of GS in China, but the prevalence of heterozygotes in the White population is about 1% [2]. The diagnosis of clinically suspected GS relies on genetic testing [1], and to date, more than 100 different loss-of-function mutations in the SLC12A3 gene have been identified in GS patients, including missense, nonsense, frameshift, and splice site mutations, and are dispersed throughout the protein. It is mainly differentiated from Batter syndrome [3]. The primary goal of treatment is to restore electrolyte status. Individualized lifelong oral potassium or magnesium supplements or both are the main treatments for patients with GS. If oral administration is intolerable or severe hypokalemia or hypomagnesemia occurs, intravenous preparations are available [4]. Potassium-sparing diuretics may be used if persistent, symptomatic hypokalemia occurs or when continued supplementation is insufficient or the adverse effects are unacceptable [5,6].
Finerenone is a non-steroidal mineralocorticoid receptor antagonist (nsMRA) used to treat chronic kidney disease. It is an analog dihydronaphthyridine compound obtained by structural modification of dihydropyridine calcium channel blockers, with which cardiorenal protection can be achieved by inhibiting inflammation and fibrosis caused by MR activation [7]. In addition, as a potassium-sparing diuretic, it can inhibit the effects of aldosterone on multiple channels of the distal convoluted tubule and collecting duct, reduce urinary potassium excretion. The current main treatment of patients with CKD and T2DM is to reduce their risk of cardiovascular events and progression of kidney disease. Hyperkalemia is the most common adverse effect of finerenone use. In the FIGARO-DKD study, the rate of discontinuation due to hyperkalemia was 1.2%, which was higher than in the placebo group (0.4%), but there was no significant difference in the overall incidence of adverse events between the 2 groups [8].
This report describes a 35-year-old man with hypokalemia, a mass in the right adrenal gland, and a diagnosis of GS with a c.1456>A heterozygous variant of the SLC12A3 gene. Due to the development of gynecomastia because of the use of spironolactone, the treatment was switched to finerenone combined with potassium and magnesium supplementation.
Case Report
BASIC MEDICAL HISTORY:
A 35-year-old Han Chinese man was admitted to the Department of Urology, Affiliated Hospital of Qingdao University. During a health check-up, he underwent a dynamic contrast-enhanced CT scan of the lower abdomen 4 days before admission, which revealed a mass in the right adrenal gland. He had no dizziness, fatigue, nausea, vomiting, blurred vision, general weakness, or profuse sweating and was admitted to the hospital with an adrenal gland mass for surgical treatment. He was in generally good condition upon admission and did not smoke or drink. Denied any history of taking medications.
PHYSICAL EXAMINATION:
BP was 132/85 mmHg, height was 178 cm, weight was 94 kg, and BMI was 29.7 kg/m2. There was no thyroid enlargement and no obvious positive signs on cardiopulmonary and abdominal examination, and tendon reflexes and bowel sounds were normal.
AUXILIARY EXAMINATION:
Testing at admission (Table 1) revealed serum potassium 2.7 mmol/L (normal range, 3.5~5.5 mmol/L) and serum chloride 94 mmol/L (99~110 mmol/L). Due to adrenal cyst combined with hypokalemia, he was transferred to the Endocrinology Department for further testing, which showed blood gas PH 7.44 (7.35~7.45), PCO2 41.2 mmhg (35~45 mmHg), and actual base excess 3.5 mmol/L (−3~3 mmol/L). Electrolyte analysis revealed serum potassium 2.8 mmol/L (3.5~5.5 mmol/L), serum magnesium 0.58 mmol/L (0.75~1.02 mmol/L), serum chloride 96 mmol/L (99~110 mmol/L), ionized calcium 1.07 mmol/L (1.1~1.29 mmol/L), aldosterone 131.4 pg/ml (30~160 pg/ml), and renin 8.99 ng/mL/hr (0.15~2.33 ng/mL/hr). Twenty-four-hour urine electrolyte testing showed potassium 32.84 mmol/L, output 2900 mL, potassium 3.81 g/24 h, calcium 0.016 g/24 h (0.1~0.3 g/24 h), creatinine 1.84 g/24 h (0.7~1.5 g/24 h), and phosphorus 0.59 g/24 h (0.7~1.7 g/24 h). Fractional excretion of potassium was 14.79%; HbA{1c was 6.8% (3.6–6.0%), urinary microalbumin/urinary creatinine was 38.75 mg/g, urinary microalbumin was 34.6 mg/L (0–30 mg/L), anti-human insulin antibody was 18.06IU/ml (<20 IU/ml), glutamic acid decarboxylase antibody was 1 IU/ml (<30 IU/ml), C-peptide was 3.68 ng/mL (1.1~4.4 ng/mL), and INS was 16.58 uIU/mL (3.0–17.0 uIU/mL). Dynamic contrast-enhanced CT of the lower abdomen showed a nodular low-density shadow in the right adrenal region, with clear boundaries and a diameter of approximately 37 mm. No significant enhancement was noted. The possibility of a cyst was high. No obvious abnormal density was observed in the left adrenal gland. A 12-channel conventional electrocardiogram showed sinus rhythm.
GITELMAN SYNDROME DIAGNOSIS:
1) Exclude insufficient potassium intake from the digestive tract and use of thiazide diuretics or laxatives based on the patient’s medical history; 2) This patient meets the diagnostic criteria for renal potassium loss, and hypokalemia is considered to be related to increased renal potassium excretion. Diagnostic criteria for renal potassium loss are: chronic hypokalemia (<3.5 mmol/L) with inappropriate renal potassium wasting (spot potassium-creatinine ratio >2.0 mmol/mmol [>18 mmol/g], in absence of potassium-lowering drugs [1]; 3) Hypomagnesemia (<0.7 mmol/L [<1.7 mg/dl]); 4) Hypocalciuria (spot urine, calcium-creatinine ratio <0.2 mmol/mmol [<0.07mg/mg] in adults); 5) Metabolic alkalosis; 6) Increased plasma renin levels.
DNA TESTING:
According to guidelines, the diagnosis of clinically suspected GS relies on genetic testing, which should be offered to all appropriate patients [1]. SLC12A3 gene mutation is the basis for the diagnosis of Gitelman syndrome. Therefore, our patient underwent genetic testing on 13 July, 2019, showing that he carried a homozygous pathogenic variant c.1456G>A in the SLC12A3 gene (Table 2); this mutation is a missense mutation (expected to change amino acid 486 of the encoded protein from Asp to Asn). There are reports in the literature that this mutation is a hotspot mutation in Chinese patients with Gitelman syndrome [9]. Bioinformatics software predicts that it is more likely to cause disease. Taking all factors into consideration, this variant is considered to be pathogenic.
DIFFERENTIAL DIAGNOSIS:
Bartter syndrome (BS), first proposed by Bartter in 1962, is the most important genetic disorder to consider in the differential diagnosis of GS [2]. It is caused by mutations in the CLCNKB gene located in the thick ascending branch of the medullary loop. Patients often present with fatigue, fatigue, and slow growth, and laboratory tests usually show hypokalemia, metabolic alkalosis, and high levels of aldosterone, which overlap with Gitelman syndrome clinically and biochemically. However, BS onset occurs earlier (<3 years old), the symptoms are more severe, usually accompanied by growth retardation. Blood magnesium levels are often normal [10,11], and urine calcium levels are normal or elevated. It is currently believed that a chloride scavenging test can help differentiate between these 2 syndromes. If the hydrochlorothiazide test result is positive but the furosemide test result is negative, Batter syndrome can be clinically diagnosed. However, this method is cumbersome and can aggravate electrolyte imbalance. It will also produce many difficult-to-explain results and is inconvenient for clinical application [12,13]. Now, genetic testing can directly identify these 2 diseases. Other differential diagnoses of GS include other diseases causing hypokalemia, such as Liddle syndrome, tubulointerstitial nephritis, and renal tubular acidosis, which can be distinguished based on medical history, laboratory tests, and genetic testing. In addition, abuse of diuretics or laxatives and chronic vomiting should also be differentiated, which can mostly be excluded through medical history inquiry.
Specifically, the patient in our report has a mass in the right adrenal gland, which necessitates differentiation from adrenal mass diseases that could cause hypokalemia, such as adrenal adenoma, adrenal hyperplasia, adrenal carcinoma, and aldosterone-producing tumor. Diagnosis often requires pathological examination.
TREATMENT:
The patient underwent right adrenal mass resection on 1 August, 2019. Postoperative pathology showed a right retroperitoneal mass, consistent with an adrenal cyst (Figure 1). Since simple adrenal cysts typically do not cause abnormal hormone levels, the patient’s GS was likely unrelated to the adrenal cyst. It was only through further examinations in preparation for cyst removal surgery that GS was discovered and diagnosed. After surgery, the patient regularly took controlled-release potassium chloride tablets 500 mg bid, magnesium chloride 1000 mg bid, and spironolactone 20 mg bid. The serum potassium was maintained at 3.63~3.71 mmol/L (Table 3).
In October 2019, the patient developed male bilateral mammary gland development. Since spironolactone has an anti-androgen effect, this was considered to be an adverse effect of spironolactone. Immediately, spironolactone was stopped, and the serum potassium decreased, then was switched to potassium chloride 2000 mg tid and magnesium chloride 1000 mg bid, but this seriously reduced his quality of life. Serum potassium still could not be maintained at normal levels and was less than 3 mmol/l in repeated tests. Moreover, there was an obvious increase in nocturia. In September 2023, the patient was given the potassium-sparing diuretic finerenone 10 mg bid, and the doses of potassium and magnesium were both reduced to potassium chloride 1500 mg bid and magnesium chloride 1000 mg bid, respectively, and he has continued this treatment to date. Blood potassium levels increased and were mostly maintained at 3.48~3.8 mmol/L, and blood magnesium was maintained at 0.47~0.7 mmol/L. No obvious adverse reactions were found. In addition, the patient was treated with Semaglutide 1.0 mg qw and dapagliflozin 1 tablet qd for hypoglycemic treatment, and the blood glucose levels were well-controlled. Studies have shown that dapagliflozin, as an SGLT2 inhibitor, can increase blood magnesium levels. The mechanism may be related to the inhibition of sodium and glucose reabsorption by SGLT2 inhibitors, which can reduce urinary magnesium excretion and slightly elevate serum magnesium levels.
Discussion
This case study shows the beneficial effects of finerenone, a selective mineralocorticoid receptor antagonist, on a patient with GS, which is caused by mutations in the SLC12A3 gene on chromosome 16. The SLC12A3 gene encodes the thiazide drug-sensitive sodium-chloride cotransporter (NCC) in the distal convoluted tubule. The mutation can lead to a decrease in the absorption of sodium and chloride in the distal tubule, accompanied by an increase in the reabsorption of calcium ions due to electrochemical changes, and a decrease in the number of magnesium ion channels (TRPM6), resulting in a decrease in the reabsorption of magnesium ions [14]. Eventually, sodium-potassium exchange and sodium-hydrogen exchange increase at the collecting duct, resulting in increased urinary potassium and magnesium excretion, and decreased urinary calcium excretion, leading to hypokalemia, hypomagnesemia, hypocalciuria, and hypochloremic metabolism alkalosis. At the same time, due to reduced blood volume, it promotes the activation of the renin-angiotensin-aldosterone system, thereby aggravating hypokalemia and metabolic alkalosis [1]. The severity of symptoms varies widely among patients with GS. Some patients are asymptomatic or experience only mild weakness, while others exhibit severe neuromuscular symptoms such as muscle weakness, paresthesia, spasticity, and tetany or paralysis[15]. Studies show that 14% to 16% of GS patients have abnormal glucose metabolism [16,17]. Yuan et al [16] found that 53.6% of GS patients had abnormal glucose metabolism, including impaired fasting blood glucose (3.6%), impaired glucose tolerance (28.6%), type 2 diabetes (21.4%), and insulin resistance. The possible mechanism is that hypokalemia reduces insulin secretion [18]. Pancreatic insulin release is controlled by ATP-sensitive potassium channels and L-type calcium channels on the surface of b cells [19]. Hypokalemia can prevent the closure of these channels [20]. In addition, it has been found in many studies that magnesium deficiency is also related to diabetes [21,22], which can damage the insulin signaling pathway and reduce insulin sensitivity to glucose. Epidemiological studies report that about 6% of GS patients have hypokalemic paralysis, and this symptom is more common in Asian patients. In a questionnaire, increased thirst and craving for salt were prominent symptoms in 75% of GS patients, usually starting in childhood and continuing into adulthood [23]. Many symptoms are related to electrolyte abnormalities caused by the disease, but not all patients with severe electrolyte abnormalities develop clinical symptoms and vice versa. Some patients with severe hypomagnesemia are asymptomatic, whereas others with mild hypokalemia develop paralysis or cardiac arrhythmias [24]. Although the prevalence of GS in China is unknown, Liu et al [17] conducted a genetic study on a small sample of healthy Chinese people and found that the mutation frequency of SLC12A3 is about 3%, which to a certain extent can explain the high prevalence of GS in China.
The case in this report started in adulthood and was complicated by abnormal glucose metabolism, dry mouth, polydipsia, and increased nocturia. Laboratory tests showed hypokalemia, hypomagnesemia, hypochloremic metabolic alkalosis, hypocalciuria, and renin activity elevated, clinically considered to be GS. It should be distinguished from Bartter syndrome. There are similarities in biochemical changes between the 2 syndromes, which can be discriminated from the age of onset, hypomagnesemia, and growth retardation. The gene mutation site is the main basis for the diagnosis of the disease. One study found that c.179C>T is currently the most common mutation in Chinese GS patients [25]. This mutation is different from the mutation spectrum in foreign countries: IVS9+1G>T is the most common variant reported in Europe, and c.2573T>A and c.1924C>T are the most common variants found in Japan [26] (Table 4). Our patient’s gene exome sequencing showed a homozygous mutation in exon 12 c.1456G>A (p.Asp486Asn) of chromosome 16q13, which is a missense mutation and this variant has been reported in ClinVar as pathogenic.
Since GS is caused by genetic mutation, there is no radical cure so far. Treatment is mainly focused on eliminating symptoms and correcting electrolyte imbalances. Most methods include adjusting diet and supplementing potassium and magnesium (Table 5). Potassium-sparing diuretics, angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor blocker (ARB), and non-steroidal anti-inflammatory drugs (NSAIDS) are used to reduce arrhythmias, impaired glucose metabolism [27], and chondrocalcinosis that may be caused by low potassium and low magnesium. Although these medications are not necessary, according to the guidelines, in cases of persistent, symptomatic hypokalemia where supplements are insufficient or side effects are unacceptable, or both, these classes of drugs can be used. Oral potassium supplementation is generally used, and potassium chloride is recommended because other potassium salts combine with difficult-to-absorb anions such as gluconic acid or aspartic acid, which cannot correct hypokalemia and may even aggravate metabolic alkalosis [15]. For magnesium supplementation, it is recommended to use organic acid salt preparations (aspartate, citrate), which have higher bioavailability. Taking them with meals can alleviate gastrointestinal symptoms. It is generally believed that the correction target for serum potassium in GS patients is above 3.0mmol/L, and serum magnesium is above 0.6mmol/L [1]. There is consensus that when there is refractory electrolyte imbalance or treatment requires intravenous potassium and magnesium supplementation, potassium-sparing diuretics can be used in combination. Potassium-sparing diuretics mainly include non-aldosterone antagonist drugs and aldosterone antagonist drugs. Non-aldosterone antagonists can directly antagonize epithelial sodium channels and reduce potassium excretion. Aldosterone can bind to the mineralocorticoid receptor (MR) in distal renal tubular epithelial cells to form an aldosterone-MR complex [28], preserving Na+ and excreting K+. Mineralocorticoid receptor antagonists (MRAs) can be divided into traditional steroids (such as spironolactone and eplerenone) and new-generation non-steroidal drugs (such as finerenone), which inhibit aldosterone on the distal convoluted tubule. And the role of multiple channels in the collecting duct to reduce urinary potassium excretion. In addition, there have been isolated reports of the use of renin-angiotensin system inhibitors for the treatment of GS[29], which can exacerbate renal sodium wasting and increase the risk of hypovolemia; Non-steroidal anti-inflammatory drugs (NSAIDs) such as indomethacin, although indicated in guidelines for GS, have significant gastrointestinal side effects, and long-term use can cause cardiovascular side effects. The latter 2 drugs are not considered in the treatment of this patient because they are not routinely used and have significant side effects.
Spironolactone is a targeted potassium-sparing diuretic and its steroidal structure allows it to bind to the MR ligand domain and exert an anti-MR effect. It has a strong antagonistic effect on MR, but due to its poor selectivity for receptors, spironolactone has similarities with sex hormones in molecular structure, so it can not only bind to MR, but also to androgen receptor/progesterone receptor, showing sex hormone-related adverse reactions; the most common ones are gynecomastia [30], breast pain or tenderness, and erectile dysfunction [28]. Premenopausal women may experience menstrual disorders, breast hyperplasia, and pain [31]. Compared with spironolactone, eplerenone has a significantly lower affinity for androgen and progesterone receptors without corresponding adverse effects, but these drugs can exacerbate renal salt depletion and cause hypotension. Amiloride and triamterene are sodium channel blockers that reduce sodium reabsorption and retain potassium by blocking ENaC. In situations where potassium retention is needed, they can serve as alternatives to spironolactone. However, these medications are not available in our local area. As a non-steroidal MRA, finerenone is a naphthyridine derivative developed based on the dihydropyridine (DHP) structure. It binds to MR more completely and has stronger MR antagonistic effect [32]. In addition, its affinity for androgen and progesterone receptors is extremely low, so there are no sex hormone-related adverse reactions. In the FIDELIO study [33] and FIGARO [8] study, no cases of breast hyperplasia were reported in the finerenone group, and the incidence of gynecomastia was similar to that in the placebo group, with neither exceeding 0.2%. As an MRA, finerenone can competitively bind to MR with aldosterone, promoting MR degradation or interfering with the formation of hormone-receptor complexes, thereby exerting an antagonistic effect and reducing the risk of inflammation and fibrosis. It can also significantly reduce the level of protein in the urine, which could produce a protective effect on the heart and kidneys. The related mechanism may be the following. In the kidneys, MRA can inhibit the expression of inflammatory transmitters and inflammatory factors and reduce collagen deposition and macrophage infiltration in nephrons [34]. In blood vessels and the heart, it can increase the activity of superoxide dismutase, enhance the bioavailability of nitric oxide, and reduce the level of superoxide anions, helping to alleviate vascular endothelial dysfunction [35]. Therefore, finerenone is currently mainly used to reduce cardiac and renal risks in patients with T2DM.
The 2022 American Heart Association (AHA) Guidelines, the 2022 Guidelines published by the Kidney Disease Improving Global Outcomes Organization (KDIGO), and the 2023 American Diabetes Association (ADA) Guidelines all state that finerenone can reduce the risk of adverse cardiorenal events in patients with diabetes combined with CKD on the basis of conventional antidiabetic treatment. Due to our patient’s concurrent conditions of GS and type 2 diabetes, along with elevated levels of urinary microalbumin and kidney dysfunction, it was ultimately decided to treat with finerenone, which proved to be effective. There are currently no reports of the use of finerenone in the treatment of Gitelman syndrome. To the best of our knowledge, this is the first case such case study.
As an aldosterone receptor antagonist, finerenone can reduce urinary potassium excretion by inhibiting the effects of aldosterone on multiple channels of the distal convoluted tubule and collecting duct. Therefore, the most common adverse effect of finerenone is hyperkalemia, but the risk is relatively low. In the FIDELIO-DKD study, the incidence of hyperkalemia-related adverse events in the finerenone group was higher than that in the control group, but there were no fatal hyperkalemia adverse events [33]. In the FIGARO-DKD trial, the rate of discontinuation of treatment due to hyperkalemia was higher in the finerenone group than in the placebo group, but there was no significant difference in the overall incidence of adverse events [8]. In this case, the patient developed bilateral gynecomastia after using spironolactone for 2 months, which was considered an adverse effect of spironolactone. After stopping spironolactone, the serum potassium level dropped lower than before and could not be maintained at normal levels. Finerenone was added, and the serum potassium level rose to higher than before, and no relevant adverse reactions have occurred so far.
Conclusions
For Gitelman syndrome patients who cannot tolerate non-selective mineralocorticoid receptor antagonists, especially male patients, finerenone can be considered to assist in potassium-sparing and magnesium-replenishing treatment. This provides more options for the future treatment of GS patients, reduces drug adverse effects and dosage, and will further reduce patients’ symptoms and improve their prognoses.
Tables
Table 1.. Important laboratory findings. Table 2.. Results of exon sequencing of SLC12A3 gene.
Table 2.. Results of exon sequencing of SLC12A3 gene. Table 3.. Treatment process and results.
Table 3.. Treatment process and results.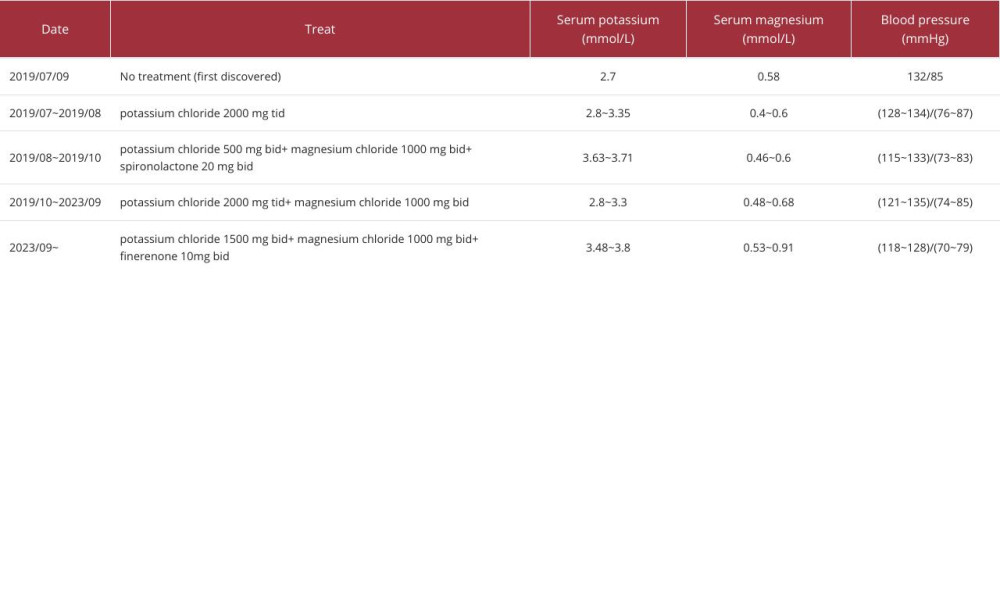 Table 4.. Number of reports on common GS gene variants in China, Japan, and Europe.
Table 4.. Number of reports on common GS gene variants in China, Japan, and Europe.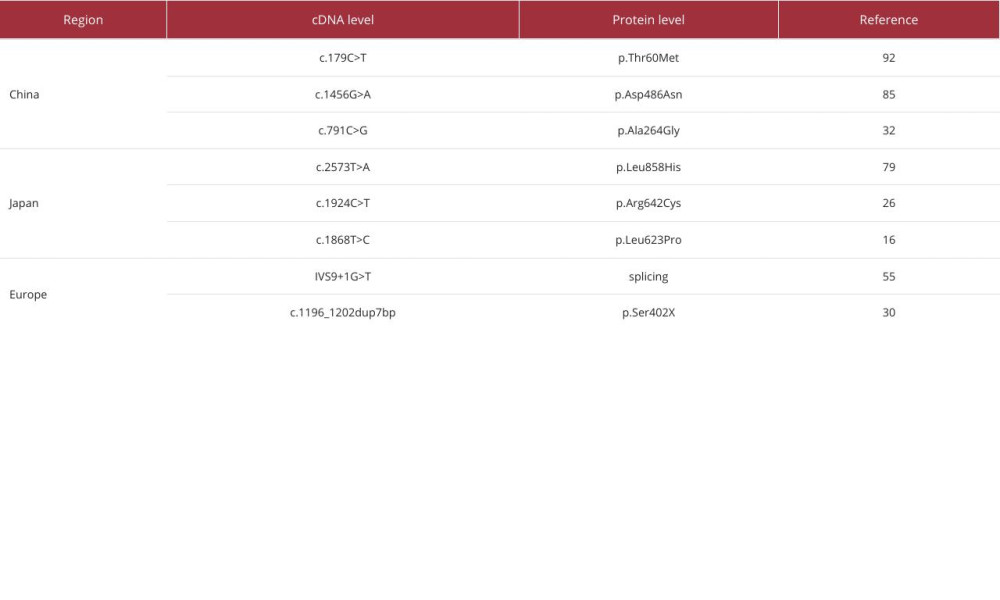 Table 5.. Recommended methods, mechanism, adverse effects, and applications.
Table 5.. Recommended methods, mechanism, adverse effects, and applications.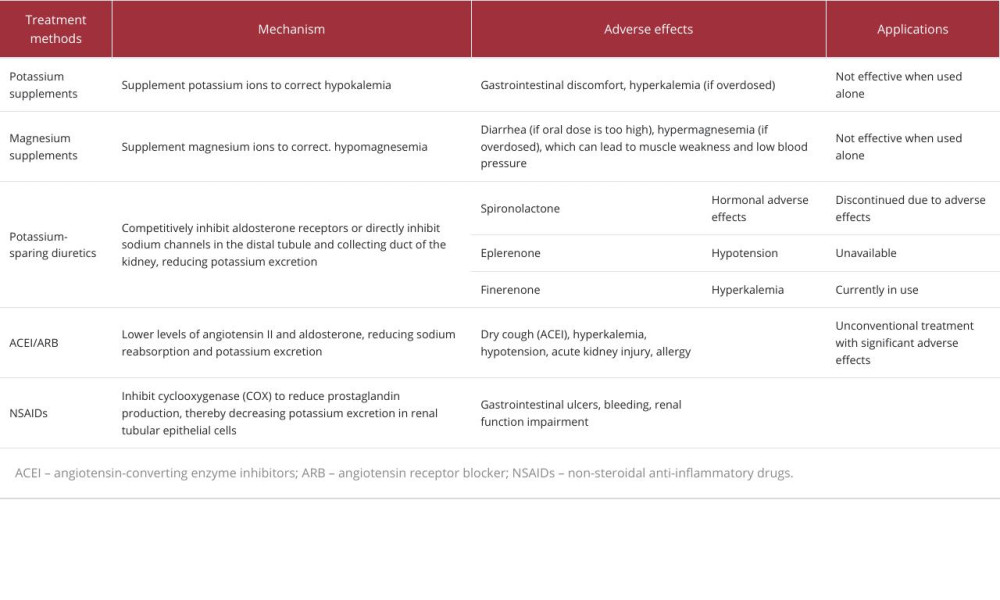
References:
1.. Blanchard A, Bockenhauer D, Bolignano D, Gitelman syndrome: Consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference: Kidney Int, 2017; 91(1); 24-33
2.. Knoers NVAM, Levtchenko EN, Gitelman syndrome: Orphanet J Rare Dis, 2008; 3; 22
3.. Nuñez-Gonzalez L, Carrera N, Garcia-Gonzalez MA, Molecular basis, diagnostic challenges and therapeutic approaches of bartter and gitelman syndromes: A primer for clinicians: Int J Mol Sci, 2021; 22(21); 11414
4.. Ito Y, Yoshida M, Nakayama M, Eplerenone improved hypokalemia in a patient with Gitelman’s syndrome: Intern Med, 2012; 51(1); 83-86
5.. Colussi G, Rombol G, De Ferrari ME, Correction of hypokalemia with anti-aldosterone therapy in Gitelman syndrome: Am J Nephrol, 1994; 14(2); 127-35
6.. Heinig R, Gerisch M, Engelen A, Pharmacokinetics of the novel, selective, non-steroidal mineralocorticoid receptor antagonist finerenone in healthy volunteers: Results from an absolute bioavailability study and drug–drug interaction studies in vitro and in vivo: Eur J Drug Metab Pharmacokinet, 2018; 43(6); 715-27
7.. Capelli I, Gasperoni L, Ruggeri M, New mineralocorticoid receptor antagonists: Update on their use in chronic kidney disease and heart failure: J Nephrol, 2020; 33(1); 37-48
8.. Pitt B, Filippatos G, Agarwal R, Cardiovascular events with finerenone in kidney disease and type 2 diabetes: N Engl J Med, 2021; 385(24); 2252-63
9.. Mou L, Tang M, Zhu L, Spectrum of variants in a large Chinese Gitelman syndrome cohort: Clin Genet, 2023; 104(6); 674-78
10.. Fukuyama S, Hiramatsu M, Akagi M, Novel mutations of the chloride channel Kb gene in two Japanese patients clinically diagnosed as Bartter syndrome with hypocalciuria: J Clin Endocrinol Metab, 2004; 89(11); 5847-50
11.. Zelikovic I, Szargel R, Hawash A, A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes: Kidney Int, 2003; 63(1); 24-32
12.. Kageyama K, Terui K, Shoji M, Diagnosis of a case of Gitelman’s syndrome based on renal clearance studies and gene analysis of a novel mutation of the thiazide-sensitive Na-Cl cotransporter: J Endocrinol Invest, 2005; 28(9); 822-26
13.. Mukhopadhyay S, Jana S, Chatterjee A, Quadriparesis due to Gitelman’s syndrome diagnosed with thiazide diuretic test response: Saudi J Kidney Dis Transpl, 2016; 27(2); 407-10
14.. Nijenhuis T, Vallon V, Van Der Kemp AWCM, Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia: J Clin Invest, 2005; 115(6); 1651-58
15.. Graziani G, Fedeli C, Moroni L, Gitelman syndrome: Pathophysiological and clinical aspects: QJM, 2010; 103(10); 741-48
16.. Yuan T, Jiang L, Chen C, Glucose tolerance and insulin responsiveness in Gitelman syndrome patients: Endocr Connect, 2017; 6(4); 243-52
17.. Liu T, Wang C, Lu J, Genotype/phenotype analysis in 67 Chinese patients with Gitelman’s syndrome: Am J Nephrol, 2016; 44(2); 159-68
18.. Cutler JA, Thiazide-associated glucose abnormalities: Prognosis, etiology, and prevention: is potassium balance the key?: Hypertension, 2006; 48(2); 198-200
19.. Sperling MA, ATP-sensitive potassium channels – neonatal diabetes mellitus and beyond: N Engl J Med, 2006; 355(5); 507-10
20.. Howell SL, Taylor KW, Potassium ions and the secretion of insulin by islets of Langerhans incubated in vitro: Biochem J, 1968; 108(1); 17-24
21.. Chambers EC, Heshka S, Gallagher D, Serum magnesium and type-2 diabetes in African Americans and Hispanics: A New York cohort: J Am Coll Nutr, 2006; 25(6); 509-13
22.. Van Dam RM, Hu FB, Rosenberg L, Dietary calcium and magnesium, major food sources, and risk of type 2 diabetes in U.S: Black Women. Diabetes Care, 2006; 29(10); 2238-43
23.. Cruz DN, Shaer AJ, Bia MJ, Gitelman’s syndrome revisited: An evaluation of symptoms and health-related quality of life: Kidney Int, 2001; 59(2); 710-17
24.. De Jong JC, Van Der Vliet WA, Van Den Heuvel LPWJ, Functional expression of mutations in the human NaCl cotransporter: Evidence for impaired routing mechanisms in Gitelman’s syndrome: J Am Soc Nephrol, 2002; 13(6); 1442-48
25.. Ma J, Ren H, Lin L, Genetic features of Chinese patients with Gitelman syndrome: Sixteen novel SLC12A3 mutations identified in a new cohort: Am J Nephrol, 2016; 44(2); 113-21
26.. Fujimura J, Nozu K, Yamamura T, Clinical and genetic characteristics in patients with Gitelman syndrome: Kidney Int Rep, 2018; 4(1); 119-25
27.. Tseng M-H, Yang S-S, Hsu Y-J, Genotype, phenotype, and follow-up in Taiwanese patients with salt-losing tubulopathy associated with SLC12A3 mutation: J Clin Endocrinol Metab, 2012; 97(8); E1478-82
28.. , Multidisciplinary expert consensus for clinical application of mineralocorticoid receptor antagonists in China.: Zhonghua Nei Ke Za Zhi, 2022; 61(9); 981-99
29.. Brambilla G, Perotti M, Perra S, It is never too late for a genetic disease: A case of a 79-year-old man with persistent hypokalemia: J Nephrol, 2013; 26(3); 594-98
30.. De Gasparo M, Whitebread S E, Preiswerk G, Antialdosterones: Incidence and prevention of sexual side effects: J Steroid Biochem, 1989; 32(1); 223-27
31.. Kallistratos MS, Pittaras A, Theodoulidis I, Adverse effects of mineralocorticoid receptor antagonist administration: Curr Pharm Des, 2019; 24(46); 5537-41
32.. Jaisser F, Farman N, Emerging roles of the mineralocorticoid receptor in pathology: Toward new paradigms in clinical pharmacology: Pharmacol Rev, 2016; 68(1); 49-75
33.. Bakris GL, Agarwal R, Anker SD, Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes: N Engl J Med, 2020; 383(23); 2219-29
34.. Barrera-Chimal J, Lima-Posada I, Bakris GL, Mineralocorticoid receptor antagonists in diabetic kidney disease – mechanistic and therapeutic effects: Nat Rev Nephrol, 2022; 18(1); 56-70
35.. González-Blázquez R, Somoza B, Gil-Ortega M, Finerenone attenuates endothelial dysfunction and albuminuria in a chronic kidney disease model by a reduction in oxidative stress: Front Pharmacol, 2018; 9; 1131
Tables
Table 1.. Important laboratory findings.Table 2.. Results of exon sequencing of SLC12A3 gene.Table 3.. Treatment process and results.Table 4.. Number of reports on common GS gene variants in China, Japan, and Europe.Table 5.. Recommended methods, mechanism, adverse effects, and applications.Table 1.. Important laboratory findings.Table 2.. Results of exon sequencing of SLC12A3 gene.Table 3.. Treatment process and results.Table 4.. Number of reports on common GS gene variants in China, Japan, and Europe.Table 5.. Recommended methods, mechanism, adverse effects, and applications. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952791

Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Most Viewed Current Articles
07 Dec 2021 : Case report
22,759,422
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  175,936
175,936
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,499
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,510
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133